Delonix Society’s Baramati College of Pharmacy, Baramati , Pune, Maharashtra, India 413102
One of the most important and complex phases of drug development is lead optimization (LO), which turns early "hit" molecules into promising therapeutic candidates. The goal of this iterative approach is to reduce possible toxicity while improving a molecule's potency, selectivity, pharmacokinetics (ADME), and safety. In the past, LO depended on resource-intensive, empirical techniques. However, computational methods, especially virtual screening (VS), which includes structure-based (molecular docking and dynamics) and ligand-based (QSAR, pharmacophore modeling) approaches, have completely changed the area. These technologies aid in the logical design and synthesis of novel analogues by forecasting binding affinities and interaction patterns. Recent developments in artificial intelligence and machine learning are having a significant impact on LO, despite obstacles like precisely simulating receptor flexibility and desolvation effects. Generative Therapeutics Design (GTD) and Query-based molecular optimization (QMO) are two AI/ML-driven approaches that facilitate more effective chemical space exploration, improve the precision of molecular property predictions, and speed up the development of novel chemical entities. In order to overcome previous constraints and expedite the delivery of improved, safer, and more effective therapeutic candidates, this review examines the synergistic application of classic experimental, advanced computational, and transformative AI/ML approaches.
A crucial and intricate stage of drug discovery is lead optimization (LO), which turns early "hit" molecules into promising therapeutic prospects. In order to improve the potency, selectivity, pharmacokinetics (ADME properties: Absorption, Distribution, Metabolism, Excretion), and safety of promising molecules, this iterative process improves their physicochemical and pharmacological characteristics while also addressing potential drawbacks like toxicity. Finding a molecule with the ideal ratio of these characteristics to advance into preclinical and clinical development is the ultimate objective.
Lead optimization has historically been a laborious and resource-intensive process that mostly relies on empirical techniques and trial-and-error testing. However, with the use of computer approaches and, more recently, AI and machine learning (ML) tools, the field of drug development is changing quickly. In order to find potential candidates, virtual screening (VS), which includes both ligand-based (such as QSAR and pharmacophore modeling) and structure-based techniques, is essential. In order to direct the synthesis of novel analogues, these computational methods seek to anticipate binding affinities, interaction modalities, and ADMET profiles.
Lead optimization still faces several obstacles despite notable progress, including as precisely taking into consideration receptor flexibility, desolvation effects, and the intrinsic intricacy of ligand-receptor interactions. This stage is being completely transformed by the advent of AI/ML-driven techniques like Generative Therapeutics Design (GTD) and Query-based molecular optimization (QMO), which allow for more effective chemical space exploration, more accurate molecular property prediction, and faster creation of new chemical entities. In order to produce optimal drug candidates, this study will explore the various approaches used in lead optimization, emphasizing the complementary use of cutting-edge computational tools, conventional experimental methodologies, and the revolutionary effects of artificial intelligence and machine learning.
Virtual Screening :
By using the Ligand for ligand-based high-throughput virtual screening, an internal library of FAD structural analogues was produced.[27]
Accurate ranking to drive analog synthesis and correct ligand orientation within the active site are both necessary for lead optimization by virtual screening.
Substructures found in at least 5% of CNS medications were detected using SARvision, and their relative prevalence in non-CNS medications determined whether they were classified as equally preferred, non-CNS favored, or CNS-favored (C). [28]
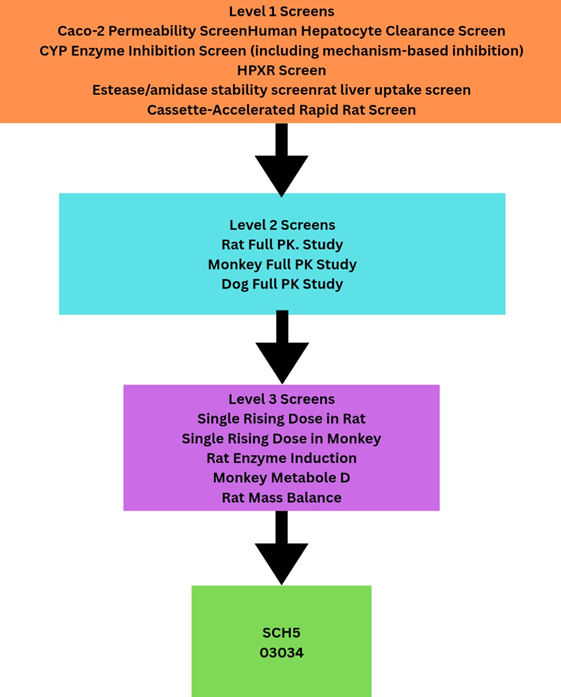
FIGURE 1. Application of the DMPK screening paradigm to the selection of HCV compounds as a component of the lead optimization and candidate selection process.
The prominent alternative to de novo design involves virtual screening of compound libraries via docking methodologies."
"Our initial docking attempts failed to yield active compounds but indicated a promising lead series with potent anti-HIV activity."
"Continuing efforts focused on the highest-ranked library compound, the inactive oxadiazole 3, which appeared to possess a suitable core."
"A subsequent virtual screening initiative demonstrated significant success."
"Notably, 11 compounds effectively inhibited protein-protein interactions in the micromolar range, with four exhibiting IC50 values under 5 µM." [20]
Steps
1. Ligand Preparation: Following geometry tests, ligands were protonated at N1 after being drawn identically in Sybyl.
2. Protein Preparation: Hydrogens were added, charges were calculated, sidechain orientations were adjusted (if necessary), and cofactors and ligands were combined to create PDB structures and homology models (such as LcDHFR).
3. Protein Minimization: The Amber force field was used to reduce the resulting protein-ligand-cofactor complexes, either completely or within a 3.5 Å radius of the ligand.
4. MD Ensemble Generation: Every 500 fs, conformational snapshots were produced via a 10,000 fs MD simulation at 300 K.
5. Definition of Active Sites and Minimization: After defining active sites (3.5 Å around the ligand/pteridine ring), the resulting MD ensemble structures were reduced using Amber. Geometry and charge verification came next.[21]
Ligand-based virtual screening:
Easy to use and efficient when ligands are known to exist; it finds similar binders but is constrained by a lack of data. De novo design is not the best option.
Essential Methods:
Structure-based virtual screening :
Limitation :
Molecular Docking and Dynamics :
Maestro v9.2 was used to preprocess the ligands and MurB reductase for molecular docking. At the allosteric site, a three-phased Glide v5.7 docking approach (VHTS, SP, XP) was employed. In order to find strong binders, the XP technique produced and ranked 10,000 poses per ligand using XPGscore, excluding those with positive scores.
To assess stability and conformational changes, MD simulations of the MurB reductase-lead1 complex were carried out using Desmond v3.0 in Maestro v9.2. Before a 5000 ps NPT simulation at 310 K, the system, which contains about 40,914 atoms, underwent multi-step reduction and brief relaxation simulations (NVT, NPT). Hydrogen bonding interactions, stability, RMSD, and RMSF were then examined. [27]
In vitro phenotypic experiments, as well as assays for enzyme inhibition and IC50 measurement, can employ N,8-dihydroxy-8-(naphthalen-2-yl) octanamide.[18]
Docking accuracy is improved by receptor flexibility.
All ligands were docked against a receptor library in Sybyl 7.2 using the Surflex-Dock docking protocol. Probes were used to determine active sites, and the software created ligands in a sequential fashion, producing 200 postures for each ligand.
Pose selection and scoring: The pose with the highest score and the conserved geometry of the 2,4-diaminopyrimidine ring was chosen as "correct." Docking scores were a weighted average of several atomic interactions and were reported in −log 10 (K d) units, where larger denotes stronger affinity.
Ensemble Averaging: It was discovered that this method was better than Boltzmann distribution averaging for receptor ensembles, therefore the individual docking scores for each ensemble member were averaged to get a single overall docking score for each ligand.
Ligands were binned by affinity, and a score of 1 was assigned if the docked ligand fell into the same or an adjacent bin based on the docking score. Improper Orientation Rate (%IO - Ligand Orientation Accuracy): This metric measures the proportion of ligands where the top-scoring pose did not have the correct 2,4-diaminopyrimidine orientation; Grouped Ranking Score (Ligand Ranking Accuracy): This metric evaluates how well docking predicts the tightest-binding ligands.[3]
Methods :
Utility of AI\ ML Methods in Lead optimization
The ability of GTD to produce pertinent and effective inhibitor concepts was greatly enhanced by the use of a 3D pharmacophore model, which represents crucial inhibitory properties.
The iterative process of GTD enabled the successful synthesis of desired substructures, such as amino pyrazines, that were first overlooked and a wider exploration of the chemical space by gradually activating restrictions.
Even with challenges, GTD successfully produced hundreds of diverse molecular ideas that fit the pharmacophore and physicochemical property models, demonstrating its potential for lead optimization. [33]
LOMAP Method
1. Design goals
2. Algorithm
3. Our implementation is in Python Simulation methods:
4. Results [23]
Methods for Safety Risk Assessment
Both quantitative (statistical) and qualitative (judgment-based) techniques are used in construction risk measurement to measure and reduce dangers. Researchers have created a number of strategies to include safety, including activity-based quantification techniques and the risk assessor model (RAM). There are two types of safety risk assessment methods: activity-based and job-based. This study focuses on the latter. The QASR framework for TCSO analysis is presented in this research.
QSAR \ QSPR Modelling :
Through developments in de novo design, virtual screening, pharmacologically significant property prediction, and protein-ligand binding affinity estimate, computational chemisthas made a substantial contribution.[20]
Drug discovery is effectively being impacted by artificial intelligence, especially machine learning techniques like QSAR, SVMs, and Random Forests. In benchmark experiments, new developments in neural networks—particularly deep learning—improve property predictions even more, surpassing traditional techniques [19]
Steps
SAR of an HIV-1 Vif-APOBEC3G Axis Inhibitor.
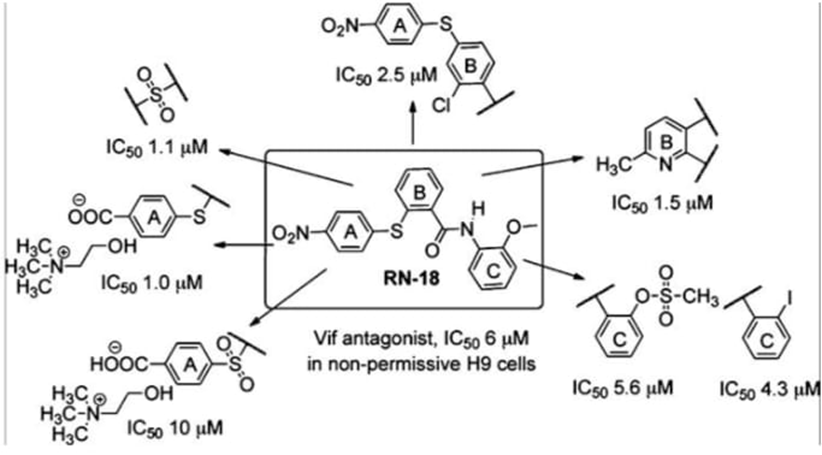
Figure 2 :SAR and lead optimization of HIV-APOBEC3G Axis inhibitor
Ring C: It is preferable to have a methyl at R1. Good activity was demonstrated by the analogues of 2-iodo (4g) and methanesulfonate (4i); methyl carboxylate (4f) at R2 also maintained action.
Bridge A-B: Other linkers (CH2 extension, amide, and urea) decreased efficacy, while sulfone (5) demonstrated five times the activity of sulfide (1).
Ring B: Methyl at position 6 (8b) increased activity, while pyridine analogues (8a, 8b) were well tolerated. During S-arylation, a novel and surprising reaction was noted.
Ring B/Bridge A-B Position: Strong analogue 11 was produced by switching the thioether attachment to para on ring B with an ortho chloro group. Compounds were rendered inactive by employing methyl/benzyl moieties or by removing ring A and bridge A-B (12).
Ring A: The water-soluble choline carboxylate (17) shown strong antiviral action, but carboxylic acid and methyl carboxylate on ring A did not. Additionally active was its oxidized sulfone analog (19).
Additional SAR concentrated on:
Ring A has a nitro group, and bridges A-B are made of sulfone. Methylene 4-mercaptobenzoate, CuI, K2CO3, SOCl2, o-anisidine, Et3N, KMnO4/MnO2, (CH3)3SnOH, and choline base were among the synthesis-related reagents.[16]
ADME
Table 1 : Propertis and their Definations
|
Terms |
Requirement |
|
Potency |
Strength the innate ability of a substance to produce the intended pharmacological action. |
|
Bioavailability |
The ability of a compound to pass through multiples barriers, such as the GI tract and the liver in order to reach the target |
|
T 1\2 |
The ability of a substance to remain in the bloodstream long enough to provide a notable pharmacological effect. |
|
Safety |
In order to develop an appropriate therapeutic index, the chemical must demonstrate adequate selectivity for its intended therapeutic action while limiting unwanted consequences. |
|
P. Acceptability |
Among other pertinent qualities, the compound should have the following suitable pharmaceutical properties: a feasible synthesis pathway, adequate water solubility, a satisfactory rate of dissolution, and strong chemical stability. |

Figure 3: Diagram illustrating the iterative process of lead optimization that results in a candidate.
AI\ Machine Learning :
Quantitative structure-activity relationship (QSAR) models are frequently created using machine learning (ML) methods, which may identify mathematical relationships between chemical characteristics and the relationship between chemical activity and property can be classified as either categorical (active, inactive, toxic, nontoxic, etc.).[11]
Conventional Approaches against AI-Powered Lead Optimization:
Traditional drug lead optimization utilizes empirical methods and is sluggish and resource-intensive, yet it works well for medications like statins and aspirin.This is being changed by AI-driven strategies that use machine learning and computational methods to speed up drug discovery. Large datasets are analyzed by these techniques in order to forecast good candidates and even suggest new chemical structures. Through virtual screening and QSAR modeling, artificial intelligence greatly broadens the scope of chemical space exploration. It lessens the need for experimental validation by assisting in the prediction of important characteristics like binding affinity and ADMET. AI also provides scalability and automation, which facilitates better decision-making and lowers expenses. Lead discovery, optimization, and even re-identifying therapeutic prospects are being successfully accomplished with the help of tools such as BIOVIA Generative Therapeutics Design (GTD). Enhancing molecular attributes such as drug-likeness and solubility is a strong suit for the Query-based molecular Optimization (QMO) framework.
Enhancing molecular attributes such as drug-likeness and solubility is a strong suit for the Query-based molecular Optimization (QMO) framework. Through protein structure prediction and the creation of new compounds, the combination of AlphaFold2 and Chemistry42 has proved crucial in the discovery of new inhibitors.[31]
ML:
These scoring functions (SFs) are evaluated using benchmarks such as the PDBbind datasets (CASF-2007, 2013, 2016), where the main metric is the Pearson correlation coefficient (Rp). Deep Neural Networks (DNNs), like DLSCORE, have been developed, and they achieve high Rp values by using a variety of descriptors. Other CNN-based SFs, like Pafnucy, and graph CNN-based SFs, like PotentialNet, offer advantages by directly generating features from crystal structures, avoiding manual feature engineering. Machine learning (ML) techniques, such as Random Forest (RF) and Gradient Boosting Decision Tree (GBDT), consistently outperform classical SFs. Hybrid SFs, which combine features from multiple SFs or integrate ML algorithms, frequently exhibit improved predictive performance. Newer SFs, like ΔvinaXGB and FFT-BP incorporate
To improve accuracy, more sophisticated elements like explicit water molecules and protein stiffness changes are incorporated into newer SFs like ΔvinaXGB and FFT-BP. Calibration on docked poses can further correct the relatively minimal influence of pose generating mistake on SF performance. Importantly, compared to classical SFs, ML-based SFs show better performance with more training data, underscoring the need of expanding datasets for future developments. [32]
CASE STUDIES :
Four Takeaways from an Effective Lead Optimization Case Study
GPCR :
Case Study 1: Identification of specific 5-HT2C agonists to treat metabolic diseases
Case study 2: Fighting obesity with a sugar-based library
Case study 3: Identification of strong and specific antagonists of the OX2 receptor[14]
The first step in target-based treatments is to identify a crucial enzyme or pathway that is ideally unique to the parasite. Following the creation of a biochemical assay in vitro, an HTS (high-throughput screen) can be used to determine which chemicals are hit.[4]
Toxic effects of lead:
Lead poisoning rarely manifests as traditional colic and constipation in affluent nations; instead, patients frequently have vague symptoms including exhaustion, joint and muscle pain, and stomach discomfort.
Treatment : use of chelating agents polyaminocarboxylic acids, of which sodium calcium edetate (EDTA) is particularly effective for lead poisoning [6]
FUTURE DIRECTION :
Present achievements and future directions
Although automation and task consolidation are the main goals of workflow optimization in the imaging department, several additional aspects should be taken into account, such as the stochastic task characteristics, human resource availability, and the particular technology being used. Workflow software, fully integrated electronic medical records, and macro-level data consolidation fueled by multi-facility networks aiming for financial and operational savings will be the main sources of future productivity increases.[2] The foundation of the combinatorial optimization model is the idea that resources and activities, like people, machines, and airplanes, are often indivisible in real-world scenarios.
Impact of Formulation: The time required to identify an optimal solution is greatly influenced by the mathematical formulation of a combinatorial optimization issue.
LP Relaxation: Although LP relaxation is a popular approximation that eliminates integrality constraints, its solution may differ significantly from the actual integer answer.
"Good" Formulation Strategies: A "good" formulation could include prioritizing identical objects, disaggregating limitations, adding limits (cutting planes), or raising variables or constraints.
Tight Bounds Are Important: Since loose bounds result in bigger coefficients and weaker LP relaxations, it is imperative to provide tight bounds for variables.
Automatic Reformulation: Most software packages now incorporate "automatic" reformulation or preprocessing processes to enhance solvability because of the crucial role that formulation plays. [24]
AL is mostly utilized for compound screening, but it can also be used to optimize hits that are found, which is a field that is ideal for investigation.[12]
RESULT
The repeated drug development process known as "lead optimization" improves "hit" molecules for safety, ADME, and potency. For directing chemical alterations, computational techniques like as molecular dynamics and virtual screening are essential, as is Structure-Activity Relationship (SAR) analysis. This method is being revolutionized by AI/ML tools, which improve property predictions and speed up chemical space research. Finding a substance that is appropriate for use in pharmaceuticals and has the best possible balance between safety and efficacy is the ultimate objective.
DISCUSSION
Lead optimization is changing dramatically, shifting from time-consuming empirical techniques to more complex computational and AI/ML-driven methodologies. Virtual screening and molecular dynamics now provide previously unheard-of efficiency and insight into molecular interactions, even though conventional methods like SAR and experimental tests are still essential. Despite persistent issues with data quality and model accuracy, the incorporation of AI/ML, especially in generative design and predictive modeling, promises to significantly speed up the development of drug candidates with optimal potency, ADME, and safety profiles. In order to navigate the diverse chemical landscape and provide effective medicines more quickly, this synergistic strategy is crucial.
ACKNOWLEDGMENT
The authors would like to express their gratitude to Prof. A. R. Shitole, Principal of the Delonix Society Baramati College of Pharmacy, for his unwavering support during the writing of this work, as well as to the college for its excellent facilities.
REFERENCES


S. D. Chavan, D. D. Chede, A. R. Shitole, Lead Optimization: Integrating Experimental, Computational, and AI/ML Approaches, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 309-319. https://doi.org/10.5281/zenodo.16737592
 10.5281/zenodo.16737592
10.5281/zenodo.16737592
