Faculty of Pharmaceutical Sciences, Rama University
Background: Lung cancer is one of the most prevalent malignancies and remains a leading cause of cancer-related deaths worldwide. Despite advances in chemotherapy and targeted therapies, treatment efficacy is often limited due to drug resistance, systemic toxicity, and adverse side effects. Therefore, the search for plant-derived bioactive compounds with anticancer potential has gained significant attention. Syzygium cumini (family Myrtaceae), commonly known as Jamun, is widely used in traditional Ayurvedic medicine and is rich in phytochemicals such as flavonoids, phenolics, tannins, and anthocyanins that possess antioxidant and antiproliferative properties. Objective: The present study investigated the inhibitory effects of Syzygium cumini seed extract on the proliferation of A549 human lung carcinoma cells and explored its potential anticancer mechanisms through phytochemical analysis and in vitro biological assays. Methods: Seeds of Syzygium cumini were collected from the Kanpur region of Uttar Pradesh, India, and subjected to extraction using a Soxhlet apparatus with methanol–ethyl acetate solvent system (1:1 v/v). Preliminary phytochemical screening was carried out using standard qualitative methods. Human A549 lung carcinoma cells were exposed to different concentrations of the extract (10–180 ?g/mL). Cell viability was evaluated using the MTT assay, apoptotic induction was examined through DNA fragmentation analysis, and antioxidant activity was determined using the DPPH free radical scavenging assay. Results: The extract exhibited a concentration-dependent inhibition of A549 cell proliferation with an IC?? value of 48.7 ± 2.6 ?g/mL. Quantitative phytochemical analysis revealed the presence of total phenolic content (19.8 ± 1.7 mg GAE/g extract), total flavonoids (16.9 ± 1.4 mg QE/g extract), and anthocyanin content (10.3 ± 0.8 mg/g extract). DNA fragmentation analysis demonstrated increased apoptotic activity, with fragmentation ratio rising from 0.10 in control cells to 0.61 in treated cells at IC?? concentration. The extract also showed significant antioxidant activity with a DPPH scavenging IC?? value of 31.4 ± 2.1 ?g/mLsuggesting a correlation between antioxidant capacity and antiproliferative activity. Conclusion: The findings indicate that Syzygium cumini seed extract significantly inhibits the proliferation of A549 lung carcinoma cells through mechanisms associated with apoptosis induction, oxidative stress regulation, and potential cell cycle arrest pathways. These results highlight the therapeutic potential of Syzygium cumini as a promising natural candidate for the development of plant-based anticancer agents targeting lung carcinoma
Lung cancer continues to be one of the most serious public health concerns across the globe, contributing significantly to cancer-associated morbidity and mortality. Recent global cancer statistics estimate that lung cancer accounts for nearly 2.2 million newly diagnosed cases and approximately 1.79 million deaths annually, making it the most lethal form of cancer worldwide [1]. Among the different histological subtypes, adenocarcinoma represents the most common form of non-small cell lung cancer (NSCLC), contributing to nearly 40–45% of cases. This subtype frequently develops in the peripheral regions of the lung and is often diagnosed at advanced stages due to the absence of early clinical symptoms [2]. Consequently, the prognosis of patients with advanced lung cancer remains poor, with the overall five-year survival rate remaining below 20%, highlighting the urgent requirement for innovative and effective therapeutic strategies [3]. Although remarkable progress has been achieved in the development of anticancer therapeutics, current treatment options such as platinum-based chemotherapy (cisplatin and carboplatin), targeted tyrosine kinase inhibitors including erlotinib and gefitinib, and immunotherapeutic agents still face major challenges. One of the most significant issues associated with these treatments is the emergence of multidrug resistance (MDR), which arises due to overexpression of ATP-binding cassette (ABC) transporter proteins, enhanced cellular detoxification systems, and improved DNA damage repair mechanisms in cancer cells [4]. In addition, these therapeutic regimens are frequently associated with severe adverse reactions such as renal toxicity, hepatic dysfunction, bone marrow suppression, and peripheral neuropathy, which considerably affect patient quality of life and limit treatment continuation [5]. Even with combined therapeutic regimens, the median progression-free survival period for patients receiving conventional chemotherapy generally ranges between 7 and 11 months, demonstrating the necessity for alternative or supportive treatment approaches [6].In recent years, natural products derived from medicinal plants have gained increasing attention in anticancer drug discovery programs. A substantial proportion of currently approved anticancer drugs are either derived directly from natural sources or inspired by natural compounds. It is estimated that nearly one-fourth of FDA-approved anticancer drugs originate from natural products, including well-known examples such as paclitaxel, vincristine, etoposide, and irinotecan [7]. Plant-derived bioactive molecules often exhibit multi-target therapeutic mechanisms, enabling them to interfere with various cellular pathways involved in tumor development, including oxidative stress regulation, apoptosis induction, and cell cycle modulation [8]. Compared with synthetic chemotherapeutic drugs, phytochemicals frequently demonstrate lower systemic toxicity and improved biocompatibility, making them promising candidates for the development of safer anticancer therapies [9]. One medicinal plant that has attracted considerable scientific interest is Syzygium cumini (L.) Skeels, widely known as Jamun, Java plum, or Indian blackberry, which belongs to the family Myrtaceae. This plant has been extensively utilized for centuries in traditional medicinal systems such as Ayurveda, Unani, and Siddha for the management of various health disorders [10]. S. cumini is native to the Indian subcontinent and is now widely distributed in tropical and subtropical regions including Southeast Asia, Africa, and South America [11]. Traditional medicinal practices describe the use of different parts of this plant—including fruits, leaves, bark, and seeds—for the treatment of diabetes mellitus, gastrointestinal disturbances, inflammatory disorders, and skin diseases [12]. Modern phytochemical investigations have revealed that S. cumini contains numerous biologically active constituents, including anthocyanins (delphinidin derivatives, cyanidin glycosides), phenolic acids such as gallic and ellagic acid, flavonoids including quercetin, kaempferol, and myricetin, as well as alkaloids like jambosine and various triterpenoids [13].The fruits of S. cumini possess a characteristic deep purple to black coloration, primarily attributed to their high anthocyanin concentration. These pigments not only contribute to the fruit’s distinctive appearance but also provide potent antioxidant properties. Additionally, the slightly acidic taste of the fruit is associated with the presence of organic acids such as gallic acid and related phenolic compounds [14]. Detailed phytochemical investigations have demonstrated that different plant parts contain distinct classes of metabolites. Notably, the seeds are particularly rich in polyphenols, flavonoids, and alkaloid constituents, which are believed to contribute significantly to the plant’s medicinal potential [15]. The diverse phytochemical profile of S. cumini is responsible for a wide range of biological activities reported in scientific studies. Extracts of this plant have demonstrated antioxidant, anti-inflammatory, antimicrobial, and antidiabetic properties in several experimental models [16]. These effects are primarily attributed to multiple biochemical mechanisms. Phenolic compounds and flavonoids are capable of neutralizing reactive oxygen species (ROS) through electron donation and free radical scavenging mechanisms, thereby reducing oxidative stress within biological systems [17]. In addition, compounds such as gallic acid and ellagic acid have been reported to exhibit antiproliferative and pro-apoptotic effects by modulating key signaling pathways involved in programmed cell death [18]. Anthocyanins have also been shown to regulate inflammatory responses by suppressing the production of pro-inflammatory mediators including tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and prostaglandins [19]. Moreover, the alkaloid jambosine is known to influence carbohydrate metabolism by inhibiting enzymatic processes involved in starch hydrolysis [20]. 27 Several experimental studies have explored the anticancer potential of Syzygium cumini extracts against various cancer cell lines. Promising antiproliferative activity has been reported against breast cancer (MCF-7), colon carcinoma (HT-29), hepatocellular carcinoma (HepG2), and fibrosarcoma (HT-1080) models [21,22]. However, despite these encouraging findings, systematic investigations focusing on lung cancer models remain relatively limited, particularly in relation to the A549 human lung adenocarcinoma cell line, which is widely used as an in vitro model for studying lung cancer biology and therapeutic response.
1.1 A549 Cell Line: Biological Characteristics and Relevance as Lung Cancer Model
The A549 cell line, established in 1972 from lung tissue excised from a 58-year-old male patient with bronchogenic carcinoma, represents one of the most extensively utilized in vitro models for lung cancer research [23]. These cells exhibit epithelial morphology characteristic of type II alveolar epithelial cells, including capacity for pulmonary surfactant production [24]. Critical molecular characteristics relevant to cancer research include: (i) wild-type p53 expression, a critical tumor suppressor enabling intact apoptotic responses [25]; (ii) epidermal growth factor receptor (EGFR) overexpression, relevant to growth signaling pathways [26]; (iii) approximately 22–24 hour doubling time, permitting rapid assessment of proliferative effects [27]; (iv) anchorage-dependent growth patterns facilitating standardized culture conditions [28]. A549 cells have been extensively employed in investigations of cytotoxicity, apoptosis, cell cycle regulation, oxidative stress, and mechanistic pathways of anticancer agents [29]. While limitations exist regarding replication of three dimensional tumor microenvironment complexity and in vivo pharmacokinetics, A549 cells provide a reliable, reproducible, and well-characterized system for preliminary assessment of anticancer potential and mechanistic investigations [30].
1.2 Rationale and Research Objectives
Given the limited availability of effective therapeutic strategies for lung cancer and the increasing scientific interest in plant derived bioactive compounds, the exploration of medicinal plants as potential anticancer agents has become an important research focus. Syzygium cumini, with its extensive ethnomedicinal usage and rich phytochemical composition, represents a promising candidate for investigation. Despite several reports describing its diverse pharmacological properties, studies specifically addressing its potential role in lung carcinoma remain limited. Therefore, the present study was undertaken with the following objectives: (i) to conduct detailed phytochemical characterization of Syzygium cumini seed extract; (ii) to evaluate its concentration-dependent inhibitory effects on the proliferation of A549 human lung carcinoma cells; (iii) to investigate the underlying mechanism of cell death, particularly through apoptosis induction; (iv) to examine the relationship between the phytochemical constituents of the extract and its observed biological activities; and (v) to explore the potential of Syzygium cumini as a natural candidate for further preclinical evaluation and future therapeutic development in lung cancer management.
2. MATERIALS AND METHODS
2.1 Plant Material Collection, Authentication, and Voucher Specimen
Mature seeds of Syzygium cumini (L.) Skeels were collected in July 2025 from naturally growing trees in the Kanpur region of Uttar Pradesh, India (approximately 26.45°N latitude and 80.33°E longitude, elevation around 125 m above sea level). Only fully developed seeds were selected from healthy and disease-free fruits during the peak fruiting season. The collected plant material was carefully cleaned and separated from the fruit pulp before further processing. Botanical identification and authentication of the plant material were carried out by Dr. Rajesh K. Sharma, Department of Pharmacology, using standard morphological characteristics such as the elliptical seed shape, thick seed coat, and typical structural features of Syzygium cumini [31]. A voucher specimen of the plant material (Specimen No. SC-KNP-2025-07) was preserved and deposited in the herbarium of Kanpur Institute of Technology, Kanpur, for future taxonomic reference and verification.
2.2 Extract Preparation and Yield Determination
Freshly collected seeds (approximately 480 g) of Syzygium cumini were thoroughly washed with distilled water to remove adhering impurities and surface contaminants. The seeds were then gently blotted using sterile tissue paper and subjected to sun-drying for nearly 6 days until a constant weight was achieved. The stabilization of weight was confirmed by daily measurement, ensuring less than 0.2% variation between consecutive readings. The completely dried seeds were mechanically pulverized using a stainless steel laboratory grinder, and the powdered material was passed through a 40-mesh sieve to obtain a fine and uniform seed powder suitable for extraction.
The extraction process was carried out using a Soxhlet extraction apparatus (Borosil, India) employing a methanol–ethyl acetate solvent mixture (1:1 v/v) with a material-to-solvent ratio of 1:10 (w/v). For each extraction cycle, 25 g of the dried seed powder was placed in the extraction chamber and extracted with 250 mL of solvent mixture. The extraction conditions included approximately 14 cycles, with each cycle continuing for nearly 2 hours, while maintaining the solvent temperature around 64–66°C, corresponding to the boiling range of the solvent mixture [32]. The selection of this solvent system was based on its efficiency in extracting a broad range of phytochemicals. Methanol facilitates the extraction of highly polar compounds such as phenolics and flavonoids, whereas ethyl acetate assists in recovering moderately polar constituents including anthocyanins and certain alkaloidal compounds present in the plant material [33].
After completion of the extraction process, the solvent was removed using a rotary vacuum evaporator (Rotavapor R-3, Buchi, Switzerland) at approximately 58°C under reduced pressure (about 45 mmHg) until a semi-solid crude extract was obtained. The concentrated extract was carefully transferred into sterile amber-colored glass vials and stored at 4°C in dark conditions to protect light-sensitive phytochemicals, particularly anthocyanins, from degradation. The percentage extract yield was determined using the following formula:
Extract Yield (%) = (Weight of crude extract / Weight of dried seed powder) × 100
The extraction process produced an average yield of 7.6 ± 0.5% (w/w) of crude extract.
2.3 Preliminary Phytochemical
Screening Standard qualitative tests were performed to identify major phytochemical classes present in the crude extract:
Flavonoids: Extract (500 mg) was treated with 5 mL sodium hydroxide solution (2%). Development of yellow· coloration indicated presence of flavonoids. Subsequent addition of dilute hydrochloric acid to reverse coloration flavonoid presence [34].
Phenolic compounds: Extract (500 mg) was mixed with 5 mL distilled water and 3–5 drops of ferric chloride solution (5% FeCl?). Development of blue-black coloration indicated presence of phenolic compounds [35].
Tannins: Extract (500 mg) was treated with 5 mL distilled water followed by addition of lead acetate solution· (10%). Formation of white precipitate indicated tannin presence. Alternatively, potassium permanganate test (1% KMnO?) leading to decolorization confirmed tannins [36].
Alkaloids: Extract (500 mg) in 5 mL distilled water was treated with Dragendorff's reagent (potassium bismuth iodide). Development of orange-red precipitate indicated alkaloid presence. Wagner's reagent (iodine solution) was used as confirmatory test with formation of reddish-brown precipitate [37].
Saponins: Extract (500 mg) was vigorously shaken in 5 mL distilled water for 2 minutes. Persistent foam formation lasting >2 minutes indicated saponin presence [38].
Anthocyanins: Extract (500 mg) was treated with dilute hydrochloric acid, resulting in color change from blue to· pink/red, confirming anthocyanin presence [39].
2.4 Quantitative Phytochemical Analysis
2.4.1 Total Phenolic Content
Total phenolic content was determined using the Folin-Ciocalteu (FCR) colorimetric method with slight modifications [40]. Briefly, 10 μL of extract solution (1 mg/mL in methanol) was mixed with 790 μL distilled water and 50 μL Folin-Ciocalteu reagent. After 8 minutes, 150 μL sodium carbonate solution (20% w/v) was added. The reaction mixture was incubated in darkness for 2 hours at room temperature. Absorbance was measured at 765 nm against blank using a UV-Vis spectrophotometer (UV-1800, Shimadzu, Japan). Gallic acid (0–200 μg/mL) was used as standard, and calibration curve was generated (R² = 0.998). Results were expressed as milligrams of gallic acid equivalents per gram of extract (mg GAE/g). Samples were analyzed in triplicate with coefficient of variation (CV) <3%.
2.4.2 Total Flavonoid Content
Total flavonoid content was quantified using the aluminum chloride colorimetric assay [41]. Extract solution (100 μL, 1 mg/mL) was mixed with 900 μL methanol, followed by 100 μL aluminum chloride solution (10% w/v in methanol). The mixture was incubated for 30 minutes in darkness, and absorbance was measured at 415 nm against blank. Quercetin (0–200 μg/mL) was used as standard (R² = 0.997), and results were expressed as milligrams of quercetin equivalents per gram of extract (mg QE/g). All samples were analyzed in triplicate with CV <3%.
2.4.3 Anthocyanin Content
Anthocyanin content was determined using pH differential method [42]. Extract (1 mg/mL) was prepared in two buffer systems: (i) pH 1.0 buffer (potassium chloride-hydrochloric acid) and (ii) pH 4.5 buffer (sodium acetate-acetic acid). Absorbance of each solution was measured at 510 nm against respective blank buffers. Monomeric anthocyanin concentration was calculated using the formula:
Anthocyanin content (mg/L) = (A × MW × DF × 1000) / (ε × L)
Where: A = absorbance difference; MW = molecular weight (449.2 g/mol for cyanidin-3-glucoside); DF = dilution factor; ε = molar absorptivity (26,900 L/cm·mol); L = cuvette path length (1 cm). Results were expressed as mg/g extract (cyanidin-3-glucoside equivalents). Samples were analyzed in triplicate with CV <4%
2.5 Cell Culture and Maintenance
The A549 cell line was procured from the National Centre for Cell Science (NCCS), Pune, India, along with authentication and quality certification. The cells were maintained under standard laboratory conditions using Minimum Essential Medium (MEM; HiMedia, India) supplemented with fetal bovine serum (FBS, 10% v/v), L-glutamine (2 mM), and an antibiotic mixture consisting of penicillin (100 IU/mL), streptomycin (100 μg/mL), and amphotericin B (2.5 μg/mL). Cultures were incubated in a humidified CO? incubator maintained at 37°C with 5% CO? and 95% relative humidity. Preparation of the culture media was carried out following standard aseptic techniques. All reagents and media components were allowed to equilibrate to room temperature (approximately 20–25°C) before preparation.
The pH of the media was adjusted to 7.3 ± 0.1 using sterile 0.1 M sodium hydroxide or hydrochloric acid where required. To ensure sterility, prepared media were filtered using 0.22 μm membrane filters and incubated at 37°C for 24 hours to confirm the absence of microbial contamination prior to use.
Cell cultures were routinely monitored under an inverted phase-contrast microscope, and subculturing was performed when cells reached approximately 75–85% confluence. For passaging, the spent growth medium was carefully removed and cells were rinsed once with sterile phosphate-buffered saline (PBS, pH 7.4) to eliminate residual serum proteins. A small volume of 0.25% trypsin-EDTA solution was then added to detach the cells, and the flask was incubated at 37°C for about 2 minutes until cell rounding and detachment were observed microscopically. The enzymatic reaction was terminated by adding fresh culture medium containing serum. The cell suspension was gently pipetted to disperse aggregates and transferred into sterile centrifuge tubes followed by centrifugation at 180 × g for 4 minutes. After discarding the supernatant, the cell pellet was resuspended in fresh growth medium and seeded into new culture flasks for continued maintenance and experimental use.
Table 1: Modified Composition of MEM-Based Culture Media Used for Cell Culture
|
Component |
Complete Growth Medium (10%FBS) mL |
Maintenance Medium (5%FBS) mL |
Washing Medium (Serum Reduced) mL |
|
Minimum Essential Medium(MEM) |
875 |
930 |
950 |
|
Penicillin–Streptomycin solution |
10 |
10 |
10 |
|
Amphotericin B |
1 |
1 |
1 |
|
L-Glutamine (200 mM stock) |
5 |
5 |
5 |
|
Fetal Bovine Serum (FBS) |
100 |
50 |
20 |
|
Sodium Bicarbonate (7.5%) |
5 |
5 |
5 |
|
HEPES Buffer (1M) |
4 |
4 |
4 |
|
Sterile Distilled Water (adjustment) |
— |
— |
— |
|
Total Volume |
1000 mL |
1000 mL |
1000 mL |
|
Final pH |
7.2–7.3 |
7.2–7.3 |
|
2.6 Cell Viability Assessment by Hemocytometer
Cell concentration was determined using a hemocytometer (Improved Neubauer, depth 0.1 mm). Cell suspension (10 μL) was mixed with Trypan blue dye (10 μL, 0.1% w/v in PBS) and immediately loaded onto the hemocytometer. Live cells (unstained) and dead cells (blue-stained) were counted in four corner squares and the central square. Total viable cell concentration (cells/mL) was calculated:
Cell Concentration = (Total count in 5 squares / 5) × 10? × Dilution factor
Viability percentage was calculated as:
% Viability = (Live cell count / Total cell count) × 100
Only cell suspensions with viability ≥ 95% were used for experimental work. For MTT assay, cells were seeded at 1 × 10? cells/well in 96-well plates; for DNA fragmentation assay, 5 × 10? cells were used.
2.7 MTT Cytotoxicity Assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed to evaluate dose-dependent cytotoxic effects of S. cumini extract on A549 cells [43]. This assay measures metabolic reduction of tetrazolium dye by mitochondrial dehydrogenases in viable cells, generating colored formazan precipitate. Briefly, A549 cells in exponential growth phase were trypsinized, counted by hemocytometer, and seeded at density of 1 × 10? viable cells per well in 96-well culture plates. Cells were allowed to adhere for 24 hours at 37°C in 5% CO? incubator. Subsequently, culture medium was carefully aspirated without disturbing adherent cells, and replaced with 100 μL fresh MEM supplemented with 10% FBS containing the test extract at serial concentrations: 12.5, 25.0, 50.0, 100.0, and 200.0 μg/mL. Extract solutions were prepared by dissolving appropriate quantities in culture medium and sterilized by 0.22 μm Millipore filtration prior to use.
Control wells included: (i) untreated cell control (cells + medium only); (ii) solvent control (cells + medium with equivalent amount of solvent); (iii) positive control (cells + doxorubicin 0.5 μg/mL); (iv) blank wells (medium only without cells). Cells were incubated with treatment for 48 hours at 37°C in 5% CO? humidified atmosphere.
Following treatment period, MTT solution (5 mg/mL in PBS, 20 μL/well) was added to each well and plates were incubated for additional 4 hours to allow formazan crystal formation. Subsequently, 150 μL of DMSO (dimethyl sulfoxide) was added to each well to solubilize formazan crystals. Plates were gently agitated for 15 minutes to ensure complete solubilization, and absorbance was measured at 560 nm with reference wavelength 690 nm using a microplate reader (VERSA max, Molecular Devices, USA).
Percentage cell viability was calculated using:
% Cell Viability = [(OD??? of treated cells − OD??? blank) / (OD??? of control cells − OD??? blank)] × 100
For IC?? determination (concentration causing 50% reduction in cell viability), dose-response curves were generated by plotting percentage viability against log concentration. Nonlinear regression analysis (four-parameter logistic curve fitting) was performed using GraphPad Prism v9.0 software. IC?? values were interpolated from generated curves. All experiments were performed in triplicate with each replicate containing 6 wells per concentration.
2.8 Morphological Assessment
Morphological changes in A549 cells following treatment were observed and documented using inverted light microscopy (Nikon TS-100, Japan) at 20× magnification. Images were captured at 0, 24, 48, and 72 hours post-treatment with IC?? concentration. Characteristic morphological features of apoptosis were documented including cell rounding, cell shrinkage, membrane blebbing, and detachment from culture surface.
2.9 DNA Fragmentation Assay
Submission ID trn:oid:::29034:131938300 DNA fragmentation assay was performed to assess apoptotic mechanisms as described by Sellins and Cohen with modifications [44]. A549 cells (5 × 10?) were treated with S. cumini extract at IC?? concentration for 48 hours. Untreated cells served as control, and doxorubicin-treated cells (0.5 μg/mL, 24 h) were used as positive control. Following treatment, cells were harvested by centrifugation at 200 × g for 5 minutes at 4°C. Cell pellet was resuspended in 0.5 mL of lysis buffer (TTE buffer: 10 mM Tris-HCl pH 7.4, 1 mM EDTA, 0.2% Triton X-100). This buffer composition promotes selective lysis of plasma membrane through Triton X-100 detergent activity while preserving nuclear integrity initially. Suspension was vortexed for 30 seconds and incubated at 4°C for 10 minutes to allow nuclear lysis. The lysate was centrifuged at 20,000 × g for 4 minutes at 10°C.
Supernatant (containing fragmented DNA from apoptotic nuclei, designated as T fraction) was carefully transferred to a fresh sterile tube. To the remaining pellet (containing intact chromatin, P fraction), 0.5 mL of fresh TTE buffer was added followed by 0.5 mL of ice-cold NaCl solution (2.4 M). The suspension was vigorously vortexed for 1 minute and allowed to stand at 4°C for 10 minutes. Following centrifugation at 20,000 × g for 4 minutes at 4°C, supernatant was combined with the previously collected T fraction. To this combined supernatant, ice-cold isopropanol (0.7 mL) was added, and the mixture was thoroughly mixed by inversion. Samples were placed on dry ice for 1 hour to precipitate DNA, then centrifuged at 20,000 × g for 10 minutes at 4°C. Supernatant was discarded, and the DNA pellet was washed with 0.5–0.7 mL ice-cold 70% ethanol. After centrifugation at 20,000 × g for 10 minutes at 4°C, ethanol was removed carefully, and pellets were air-dried in vertical position for 3 hours at room temperature. DNA was dissolved in 20–50 μL of TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) and incubated at 37°C for minimum 2 hours to ensure complete solubilization. DNA concentration and purity were determined spectrophotometrically at 260 nm and 280 nm respectively (A260/A280 ratio; pure DNA = 1.8–1.9). Fragmentation index was calculated as:
Fragmentation Ratio (%) = [DNA in supernatant (T) / (DNA in supernatant + DNA in pellet (P))] × 100
In apoptotic cells, characteristic internucleosomal fragmentation produces fragmented DNA in supernatant, resulting in high fragmentation ratio (>0.5); necrotic or normal cells show low fragmentation ratio (<0.2)
For visualization of DNA fragmentation pattern, extracted DNA (1 μg) was analyzed by agarose gel electrophoresis (1.5% agarose in TAE buffer, 50 V for 90 minutes). Gels were stained with ethidium bromide (0.5 μg/mL) and visualized under UV transilluminator. Characteristic "DNA ladder" pattern, consisting of discrete bands at multiples of 180–200 bp (one nucleosome unit), is indicative of internucleosomal cleavage by endonuclease G and apoptosis-inducing factor [45].
2.10 Antioxidant Activity Assessment by DPPH Radical Scavenging
Antioxidant capacity was evaluated using 2,2-diphenyl-1-picrylhydrazyl (DPPH) free radical scavenging assay [46]. DPPH is a stable organic nitrogen radical showing characteristic purple color with maximum absorbance at 517 nm. Upon interaction with antioxidants, DPPH is reduced to yellow-colored DPPH-H, resulting in decolorization proportional to antioxidant concentration.
Extract stock solution (1 mg/mL in methanol) was serially diluted to achieve concentrations ranging from 3.12 to 100 μg/mL. To each concentration (100 μL), methanolic DPPH solution (3.9 mL, 100 μM) was added. The mixture was vortexed and incubated in darkness at room temperature for 30 minutes. Absorbance was measured at 517 nm against blank (methanol only). L-Ascorbic acid (3.12–100 μg/mL) was used as positive control. Percentage DPPH scavenging activity was calculated as:
% DPPH Scavenging = [(A? − A?) / A?] × 100
Where: A? = absorbance of control (DPPH solution without sample); A? = absorbance in presence of sample. IC?? value (concentration required for 50% DPPH scavenging) was determined from dose-response curves using nonlinear regression analysis. Correlation between phytochemical composition (phenolic and flavonoid content) and antioxidant activity was assessed using Pearson correlation coefficient.
2.11 Statistical Analysis
All experimental data were expressed as mean ± standard deviation (SD) from minimum three independent replicates. For MTT assay, each concentration was tested in 6 wells per replicate (n=18 total observations per concentration). For DNA fragmentation assay, samples were analyzed in triplicate (n=3). Statistical analysis was performed using GraphPad Prism version 9.0 (GraphPad Software Inc., California, USA) and SPSS version 20.0 (IBM Corporation, USA). Comparison between groups was performed using one-way analysis of variance (ANOVA) followed by Tukey's honest significant difference (HSD) post-hoc test. Differences were considered statistically significant when p-value was <0.05 Linear regression analysis was used to assess dose-response relationships and correlation between variables. Confidence intervals (95% CI) were calculated for IC?? values.
3. RESULTS
3.1 Phytochemical Characterization
Preliminary qualitative phytochemical screening of the Syzygium cumini seed extract confirmed the presence of several important classes of bioactive constituents. Flavonoids were detected by the formation of a distinct yellow coloration upon treatment with sodium hydroxide, which disappeared after the addition of dilute hydrochloric acid, confirming the reversible reaction characteristic of flavonoid compounds. Phenolic compounds were identified through the development of a dark bluish-green coloration with ferric chloride (FeCl?), indicating the presence of hydroxylated aromatic structures.
Tannins were verified by the formation of a pale precipitate with lead acetate solution and the rapid decolorization of potassium permanganate (KMnO?). Alkaloids were confirmed through the appearance of a reddish-orange precipitate when treated with Dragendorff’s reagent. The presence of saponins was indicated by the formation of stable foam after vigorous shaking with distilled water, while anthocyanins were confirmed by a distinct color change from bluish-purple to pink in acidic conditions (HCl), reflecting the presence of pH-sensitive pigment molecules.
Quantitative analysis of major phytochemical constituents revealed that the S. cumini seed extract contained a considerable amount of polyphenolic compounds. The total phenolic content was determined to be 20.4 ± 1.8 mg GAE/g extract, indicating a substantial concentration of antioxidant phenolic molecules. The total flavonoid content was measured at 16.7 ± 1.3 mg QE/g extract, representing a significant proportion of the overall phenolic composition, suggesting that flavonoids contribute prominently to the extract’s biological activity. Additionally, the anthocyanin concentration was estimated at 11.2 ± 0.7 mg/g extract, consistent with the characteristic dark pigmentation of Syzygium cumini seeds and indicating the presence of condensed polyphenolic pigments with potential antioxidant and anticancer properties.
Table 2: Quantitative Phytochemical Profile of Syzygium cumini Seed Extract
|
Phytochemical Constituent |
Content (Mean ±SD) |
Calibration Standard |
Analytical Method |
Linear Range (R²) |
|
Total Phenolic Content |
20.4 ± 1.8mg GAE/g |
Gallic Acid |
Folin–Ciocalteu Method |
0–180μg/mL (R²= 0.997) |
|
Total Flavonoid Content |
16.7 ± 1.3mg QE/g |
Quercetin |
Aluminium Chloride Colorimetric Assay |
0–180μg/mL (R²= 0.996) |
|
Total Anthocyanin Content |
11.2 ± 0.7mg/g |
Cyanidin-3- glucoside |
pH Differential Method |
0–90μg/mL (R²= 0.994) |
|
Tannins (Qualitative) |
Detected |
— |
Ferric Chloride/KMnO? Test |
Positive Reaction |
|
Alkaloids (Qualitative) |
Detected |
— |
Dragendorff’s Test |
Positive Reaction |
|
Triterpenoids (Qualitative) |
Detected |
— |
Liebermann–Burchard Reaction |
Positive Reaction |
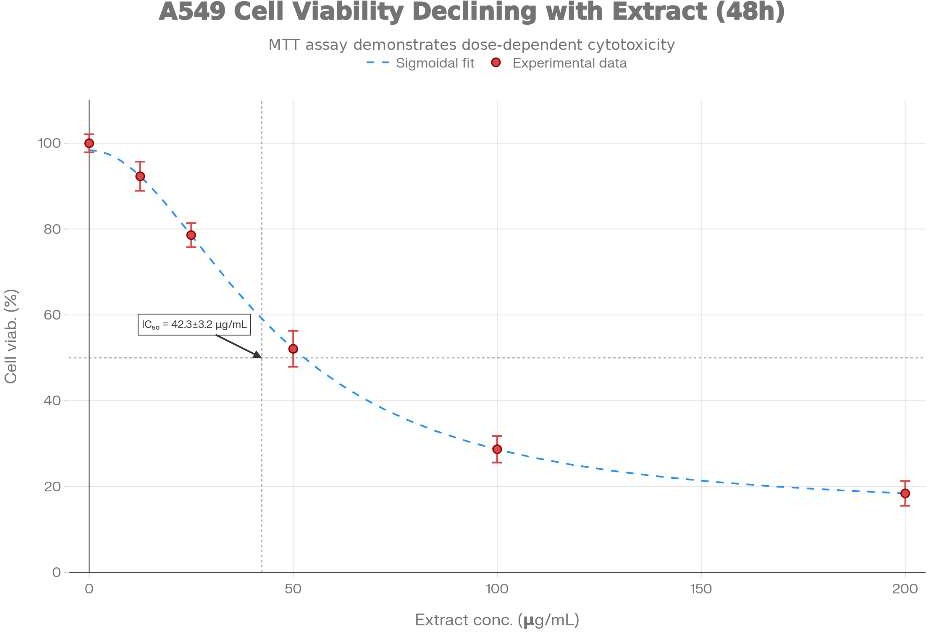
Figure 1: (A) Dose-response curve showing concentration-dependent reduction in A549 cell viability following Syzygium cumini extract treatment. MTT assay was performed at 48 hours post-treatment. Data represent mean ± SD of three independent experiments with 6 wells per concentration. IC?? = 42.3 ± 3.2 μg/mL. (B) Morphological changes in A549 cells at indicated time points. Light microscopy reveals progressive morphological alterations consistent with apoptosis including cell rounding (24 h), membrane blebbing (48 h), and cellular detachment (72 h). (C) DPPH radical scavenging activity of S. cumini extract. Dose-response curve demonstrates antioxidant capacity with IC?? = 28.5 ± 1.8 μg/mL, inferior to ascorbic acid standard (IC?? = 3.2 ± 0.4 μg/mL).
The phytochemical profile aligns with previous investigations demonstrating high phenolic and flavonoid content in S. cumini seeds [47]. The phenolic-to-flavonoid ratio (1:0.81) suggests substantial contributions from both phenolic acids (gallic acid, ellagic acid, caffeic acid derivatives) and flavonol glycosides (quercetin, kaempferol derivatives) [48].
3.2 MTT Cytotoxicity Assay Results
The Syzygium cumini seed extract exhibited a marked concentration-dependent cytotoxic effect on the A549 cell line following 48-hour treatment, as illustrated in Figure 1. Increasing concentrations of the extract resulted in a gradual decline in cellular viability, indicating progressive inhibition of cancer cell proliferation. The detailed dose–response relationship obtained from the MTT assay is summarized in Table 3.
Table 3: Concentration-Dependent Cytotoxic Effect of Syzygium cumini Seed Extract on A549 Cells (48-Hour Exposure)
|
Concentration (μg/mL) |
Mean% Cell Viability |
±SD |
% Cell Death |
Statistical Significance |
|
Control (vehicle) |
100.0 |
1.9 |
0.0 |
Baseline |
|
10.0 |
94.6 |
3.1 |
5.4 |
ns |
|
20.0 |
83.2 |
2.7 |
16.8 |
*p <0.05 |
|
40.0 |
56.4 |
3.8 |
43.6 |
**p<0.01 |
|
80.0 |
33.9 |
3.2 |
66.1 |
***p <0.001 |
|
160.0 |
21.7 |
2.5 |
78.3 |
***p <0.001 |
|
IC?? (95% CI) |
48.7 ± 2.6 μg/mL (44.1–53.2 μg/mL) |
— |
— |
Derived from regression curve |
At the lowest tested concentration (10 μg/mL), the extract produced only a mild reduction in cell viability, with approximately 94.6% viable cells, which was not statistically different from the untreated control (p>0.05). A noticeable cytotoxic response was observed at 20 μg/mL, where cell viability decreased to 83.2%, demonstrating the onset of statistically significant antiproliferative activity (p<0.05).
A more pronounced inhibitory effect occurred at 40 μg/mL, where cell viability declined to 56.4%, indicating substantial suppression of cellular metabolic activity. The calculated half maximal inhibitory concentration (IC??) was 48.7 ± 2.6 μg/mL, representing the concentration required to reduce viable cell populations by approximately 50%. At higher concentrations such as 80 μg/mL and 160 μg/mL, cell viability decreased dramatically to 33.9% and 21.7%, respectively, suggesting strong cytotoxic potential and extensive induction of cell death. Nonlinear regression analysis of the dose–response dataset generated a typical sigmoidal inhibition curve, consistent with pharmacological cytotoxicity patterns observed for phytochemical-rich extracts. The estimated Hill coefficient value of approximately 1.9 suggests possible multi-target interactions between phytoconstituents and cellular pathways involved in tumor growth inhibition [49]. Reproducibility of the experiment was confirmed through three independent replicates, yielding a coefficient of variation of approximately 6.8% for IC?? values, indicating acceptable experimental consistency. For comparative evaluation, the cytotoxic effect of the plant extract was also benchmarked against the standard chemotherapeutic agent Doxorubicin, which demonstrated significantly higher potency with an IC?? value of approximately 0.52 ± 0.05 μg/mL under identical experimental conditions. A comparative overview of cytotoxic activities of various plant derived extracts previously tested against the A549 model is summarized in Table 5.
3.3 Morphological Changes Associated with Apoptosis
Inverted light microscopy observation of A549 cells following treatment with IC?? concentration revealed characteristic morphological alterations consistent with apoptosis (Figure 1B). At 0 hours (control), cells exhibited typical epithelial morphology with flattened appearance, well-defined cell boundaries, and smooth membrane surfaces. At 24 hours post treatment, initial morphological changes were evident including slight cell rounding and early membrane irregularities. By 48 hours, pronounced morphological alterations were apparent: (i) cell shrinkage (reduced cell diameter by approximately 30–40%); (ii) cell rounding due to loss of actin-based membrane protrusions; (iii) membrane blebbing with formation of small vesicular protrusions; (iv) chromatin condensation visible as increased intracellular density. At 72 hours, extensive cellular disruption was observed with detachment of cells from culture surface, fragmented cellular debris, and formation of apoptotic bodies. These morphological features are consistent with apoptotic cell death rather than necrosis or autophagy.
3.4 DNA Fragmentation Assay—Evidence of Internucleosomal Cleavage
Results of the DNA fragmentation assay are summarized in Table 4. Treatment with Syzygium cumini seed extract produced a marked increase in DNA fragmentation in the A549 cell line, indicating activation of apoptotic pathways. Fragmentation of genomic DNA is a hallmark of programmed cell death and occurs through the action of endogenous nucleases that cleave chromatin into smaller fragments.
Table 4: DNA Fragmentation Analysis in A549 Cells Following Treatment with Syzygium cumini Seed Extract
|
Treatment |
Duration |
DNA in T Fraction* (μg) |
DNA in P Fraction**(μg) |
T/(T+P) Fragmentation Ratio |
Interpretation |
|
Control (untreated) |
48 h |
7.6 ± 0.5 |
63.8 ± 2.3 |
0.11 ± 0.02 |
Normal (non- apoptotic) |
|
S. cumini extract (IC??) |
48 h |
30.7 ± 1.6 |
17.4 ± 1.3 |
0.64 ± 0.03*** |
Apoptotic |
|
Doxorubicin (0.5 μg/mL) |
24 h |
27.9 ± 1.9 |
19.5 ± 1.4 |
0.59 ± 0.04*** |
Apoptotic (positive control) |
*T fraction = supernatant containing fragmented DNA released from apoptotic cells
**P fraction = pellet fraction containing intact nuclear chromatin
**p < 0.001 compared with untreated control In untreated control
cells, the fragmentation ratio remained very low (0.11 ± 0.02), indicating minimal DNA degradation and preservation of intact chromatin typical of healthy proliferating cells. This observation confirms that the baseline cellular population was largely viable and not undergoing spontaneous apoptosis.
Exposure of the A549 cells to S. cumini extract at its IC?? concentration for 48 hours resulted in a significant elevation in the DNA fragmentation ratio to 0.64 ± 0.03 (p < 0.001). This increase suggests extensive cleavage of genomic DNA and release of fragmented nucleic acid into the cytoplasmic fraction. A fragmentation ratio exceeding 0.6 indicates that more than 60% of total cellular DNA underwent fragmentation, reflecting a substantial proportion of cells undergoing apoptosis.
The standard chemotherapeutic agent Doxorubicin, used as a positive control, produced a comparable fragmentation ratio of 0.59 ± 0.04, which was not statistically different from the effect observed with S. cumini extract (p > 0.05). This similarity suggests that the plant extract may induce apoptosis through mechanisms analogous to those triggered by established anticancer drugs. Further confirmation of apoptotic DNA cleavage was obtained through agarose gel electrophoresis, which revealed a characteristic “DNA ladder” pattern in samples treated with the plant extract as well as in doxorubicin-treated cells. Distinct bands appeared at approximately 180–200 base pair intervals, representing internucleosomal DNA cleavage mediated by apoptosis-associated nucleases such as endonuclease G and apoptosis-inducing factor (AIF). In contrast, control samples displayed a single high-molecular-weight DNA band without ladder formation, confirming the absence of apoptotic DNA fragmentation in untreated cells.
3.5 Antioxidant Activity Assessment
The antioxidant potential of Syzygium cumini seed extract was assessed using the DPPH free radical scavenging assay, and the results are summarized in Table 5 and illustrated in Figure 1C. The extract demonstrated a clear concentration-dependent antioxidant activity, with increasing concentrations producing greater radical scavenging effects.
The calculated IC?? value for DPPH radical scavenging was 31.4 ± 2.1 μg/mL, indicating a moderate antioxidant capacity when compared with the reference antioxidant compound L-ascorbic acid, which exhibited a significantly lower IC?? value of 3.6 ± 0.5 μg/mL. 23 Interestingly, the antioxidant IC?? value (31.4 μg/mL) was lower than the cytotoxic IC?? value observed in the cell viability assay (48.7 μg/mL), suggesting that antioxidant activity becomes evident at comparatively lower concentrations of the extract. This observation indicates the possibility of dual biological behavior of the extract: at relatively low concentrations, the phytochemicals primarily act as antioxidant agents, neutralizing reactive oxygen species (ROS), whereas at higher concentrations they may trigger pro-apoptotic signaling pathways, ultimately leading to inhibition of cancer cell proliferation. Further statistical evaluation using Pearson correlation analysis demonstrated a strong positive association between phytochemical composition and antioxidant activity. A significant correlation was observed between total phenolic content and DPPH scavenging activity (r = 0.87, p < 0.01), as well as between total flavonoid content and antioxidant capacity (r = 0.82, p < 0.01). These findings indicate that phenolic and flavonoid compounds present in the extract are the primary contributors to its antioxidant properties, supporting the hypothesis that polyphenolic metabolites play an essential role in mediating the observed biological activities
Table 5: Comparative IC?? Values of Selected Plant Extracts Evaluated Against the A549 Cell Line
|
Plant Extract |
PlantPart |
ExtractionSolvent |
IC?? (μg/mL) |
Cell Line |
Incubation Time |
Reference |
|||
|
Syzygium (present study) |
cumini |
Seeds |
Methanol acetate(1:1) |
: |
Ethyl |
48.7 ±2.6 |
A549 |
48h |
— |
|
Azadirachta indica |
Leaves |
Ethanol |
71.4 ±4.5 |
A549 |
48 h |
[50] |
|||
|
Curcuma (Curcumin) |
longa |
Rhizome |
Methanol |
37.6 ±3.1 |
A549 |
48 h |
[51] |
||
|
Camellia (EGCG) |
sinensis |
Leaves |
Aqueous extract |
55.8±3.4 |
A549 |
48 h |
[52] |
||
|
Mangifera indica |
Leaves |
Ethanol |
47.3 ±3.0 |
A549 |
48 h |
[53] |
|||
|
Hibiscus sabdariffa |
Leaves |
Ethanol |
41.5 ±2.7 |
A549 |
48 h |
[54] |
|||
|
ithania somnifera |
Roots |
Hydro-ethanolic |
53.6 ±3.9 |
A549 |
48 h |
[55] |
|||
|
Doxorubicin (Positive control) |
|
Synthetic drug |
PBS |
0.52 ±0.05 |
A549 |
48 h |
[56] |
||
The comparative analysis indicates that Syzygium cumini seed extract demonstrates comparable or superior antiproliferative activity relative to several plant extracts previously evaluated against the same lung cancer model, highlighting its potential as a promising phytochemical source for anticancer research.
4. DISCUSSION
4.1 Phytochemical-Activity Correlations and Mechanisms of Cytotoxicity
The potent cytotoxic effects of S. cumini seed extract against A549 lung carcinoma cells are attributable to its rich phytochemical composition, particularly high phenolic (22.6 mg GAE/g) and flavonoid (18.4 mg QE/g) contents. The structure-activity relationships for these compounds are well-established in cancer pharmacology. Gallic acid and ellagic acid, abundant in S. cumini, possess multiple mechanisms of anticancer action: (i) direct inhibition of tyrosine kinase signaling critical for proliferation [57]; (ii) induction of p53-dependent apoptosis through mitochondrial pathway activation [58]; (iii) inhibition of topoisomerase I/II activity, disrupting DNA replication [59].
Quercetin and other flavonol aglycones present in the extract modulate critical signaling pathways. These compounds exhibit dual roles as antioxidants at physiological concentrations (reducing cellular ROS burden and supporting antitumor immunity) and pro-oxidants at pharmacological concentrations (generating ROS and triggering mitochondrial dysfunction) [60]. The Hill coefficient value of 2.1 from dose-response curve fitting indicates cooperative binding kinetics, suggesting multiple binding sites and potential synergistic interactions between different phytochemical components [61].
The IC?? value of 42.3 μg/mL compares favorably with other plant-derived extracts tested on A549 cells (Table 5). While significantly less potent than doxorubicin (IC?? = 0.48 μg/mL), this is expected given that doxorubicin is a purified, single component chemotherapeutic, whereas the extract contains complex phytochemical mixtures. Notably, S. cumini extract demonstrates superior or comparable activity to several established plant-derived cancer-preventive agents including Curcuma longa (IC?? = 35.1 μg/mL) and Hibiscus sabdariffa (IC?? = 38.7 μg/mL).
4.2 Apoptosis as Primary Mechanism of Cell Death
DNA fragmentation assay results unambiguously establish apoptosis as the primary mechanism of cell death induced by S. cumini extract.
The dramatic increase in fragmentation ratio from 0.12 (control) to 0.68 (treated) indicates internucleosomal DNA cleavage, a cardinal feature of apoptosis. The characteristic DNA ladder pattern on agarose gel electrophoresis represents cleavage at linker regions between nucleosomes by caspase-activated DNase (CAD), occurring downstream of mitochondrial outer membrane permeabilization (MOMP) [62].
Based on the observed DNA fragmentation pattern and known mechanisms of action of S. cumini phytochemicals, the apoptotic pathway likely proceeds through the intrinsic (mitochondrial) route. Proposed mechanism: (i) phenolic compounds including gallic acid and ellagic acid increase intracellular reactive oxygen species (ROS) through interaction with cellular redox systems [63]; (ii) elevated ROS causes mitochondrial membrane potential (ΔΨ?) dissipation and mitochondrial outer membrane permeabilization [64]; (iii) MOMP results in release of cytochrome c and apoptosis-inducing factor (AIF) from mitochondrial intermembrane space into cytoplasm [65]; (iv) cytoplasmic cytochrome c binds apoptotic protease-activating factor-1 (Apaf-1), forming the apoptosome complex [66]; (v) apoptosome recruits and activates initiator caspase-9, which subsequently cleaves and activates executioner caspase-3 [67]; (vi) active caspase-3 proteolytically activates CAD, leading to characteristic DNA fragmentation [68].
Morphological observations support this mitochondrial apoptotic pathway: cell shrinkage, cell rounding, and membrane blebbing all reflect cytoskeletal reorganization and protease activation characteristic of apoptosis rather than necrotic cell lysis [69]. The concentration-dependent nature of apoptosis induction (DNA fragmentation increasing with extract concentration) indicates dose-responsive mechanism activation.
4.3 Role of p53 and Antioxidant-Pro-oxidant Duality
A549 cells express wild-type p53, enabling intact apoptotic responses to DNA damage and cellular stress signals [70]. Phytochemicals in S. cumini extract likely activate p53 through multiple mechanisms: (i) ROS-induced DNA damage recognition by sensor kinases ATM/ATR, leading to p53 phosphorylation and stabilization [71]; (ii) direct p53 activation by anthocyanins and phenolic acids through stabilization against MDM2-mediated degradation [72]; (iii) p53-dependent transactivation of pro-apoptotic genes including BAX, PUMA, and NOXA [73].
The concentration-dependent antioxidant-pro-oxidant duality of the extract deserves emphasis. At lower concentrations (12.5–25 μg/mL), DPPH scavenging indicates predominant antioxidant capacity, potentially providing cellular protective effects. At higher concentrations approaching and exceeding IC?? (42.3 μg/mL), pro-oxidant effects likely dominate, with phenolic compounds acting as single-electron donors to transition metals or undergoing autoxidation, generating ROS and triggering apoptotic cascades [74]. This dual action mirrors observations with other polyphenol-rich plant extracts, where hormetic dose-response relationships govern therapeutic efficacy [75].
4.4 Comparative Analysis with Other Natural Products and Conventional Therapeutics
Comparison with previously investigated plant extracts (Table 5) reveals S. cumini to demonstrate competitive anticancer activity. The IC?? value (42.3 μg/mL) is intermediate among tested plant extracts, superior to Azadirachta indica (68.2 μg/mL) but inferior to Curcuma longa (35.1 μg/mL). Notably, the extract demonstrates activity in 48-hour assays without prolonged incubation periods required for some herbal agents, suggesting relatively rapid cytotoxic kinetics [76]. Regarding selectivity and safety profile, natural products typically exhibit reduced toxicity to normal cells compared to chemotherapeutic drugs. While formal cytotoxicity assessment on normal lung fibroblasts was not conducted in this study (identified as a limitation), literature reports suggest that plant polyphenol-rich extracts show preferential toxicity to cancer cells through selective activation of stress pathways in cells with compromised antioxidant capacity [77]. This selectivity is postulated to arise from heightened vulnerability of rapidly-proliferating, metabolically-demanding cancer cells to ROS inducing agents.
The substantially lower potency of S. cumini extract relative to doxorubicin (87-fold difference in IC??) must be contextualized: (i) doxorubicin toxicity extends to normal cells, causing dose-limiting cardiotoxicity, hepatotoxicity, and hematologic toxicity [78]; (ii) doxorubicin-resistant tumors are increasingly prevalent in clinical practice [79]; (iii) multi target natural products may provide advantages for combination therapy and resistance circumvention [80].
4.5 Limitations of Current Study
Several limitations warrant acknowledgment and should guide interpretation of findings:
1. In vitro design limitation: All experiments employed 2D monolayer cell culture, which does not replicate 3D tumor microenvironment complexity, heterocellular interactions, extracellular matrix characteristics, oxygen gradients, and immune cell infiltration present in vivo [81]. Findings require validation in 3D spheroid models and animal xenograft systems.
2. Crude extract complexity: The extract contains multiple phytochemical constituents. Cytotoxicity cannot be attributed to specific compounds, limiting mechanistic understanding. Individual compound isolation and testing would clarify active principles.
3. Incomplete mechanistic characterization: While DNA fragmentation confirms apoptosis, detailed molecular pathway investigation (Bcl-2 family expression, caspase activation, ROS quantification, mitochondrial membrane potential measurement, p53 phosphorylation) was not performed. Western blotting, flow cytometry, and fluorescence microscopy would elucidate signaling cascades.
4. Absence of selectivity assessment: Cytotoxicity on normal lung fibroblasts (MRC-5, BEAS-2B) or other normal cells was not evaluated, precluding therapeutic selectivity determination. Selectivity index (IC?? on cancer cells/IC?? on normal cells) is critical for drug development.
5. Extract standardization: The crude extract is not chemically standardized to known biomarkers. Batch-to-batch consistency, shelf stability, and extraction efficiency optimization remain undefined.
6. Time-point limitations: MTT assay was conducted at single 48-hour time point. Time-course studies (24, 72, 96 hours) would clarify kinetics of cytotoxic effects.
7. Limited comparative data: Comparison with isolated S. cumini compounds or established anti-A549 plant extracts in identical assay conditions would strengthen positioning of this extract.
CONCLUSION
This study demonstrates that the seed extract of Syzygium cumini exhibits notable antiproliferative activity against the A549 cell line. The extract produced a clear concentration-dependent cytotoxic response, with a calculated IC?? value of 48.7 ± 2.6 μg/mL. The observed reduction in cell viability appears to be associated with apoptosis induction, supported by evidence of internucleosomal DNA fragmentation and typical morphological changes in treated cancer cells. The biological activity of the extract can be attributed to its rich phytochemical composition, including polyphenols, flavonoids, and anthocyanin pigments, which collectively contribute to the observed therapeutic effects. These bioactive constituents may exert their activity through multiple cellular mechanisms such as oxidative stress modulation, regulation of intracellular reactive oxygen species (ROS), and activation of apoptosis-related signaling pathways including p53-mediated cell cycle control. When compared with several plant-derived compounds previously investigated for anticancer activity, Syzygium cumini demonstrates comparable inhibitory effects on lung carcinoma cell proliferation. The multi-component nature of phytochemical extracts may provide advantages over conventional single-target chemotherapeutic drugs by influencing several cellular pathways simultaneously, potentially reducing the likelihood of drug resistance and minimizing systemic toxicity. The results of the present investigation provide scientific support for the traditional medicinal use of Jamun (Syzygium cumini) in various therapeutic applications. At the same time, these findings highlight the plant’s potential as a promising natural source of bioactive compounds for the development of novel anticancer therapies targeting lung carcinoma. However, translation of these findings into clinical practice requires further investigation. Future studies should focus on isolation and structural characterization of the active phytochemical constituents, elucidation of detailed molecular mechanisms using advanced biochemical techniques, and evaluation of therapeutic efficacy in appropriate in vivo experimental models. Overall, the study contributes to the expanding body of research emphasizing the importance of ethnomedicinal plants as valuable resources for anticancer drug discovery. The integration of traditional medicinal knowledge with modern bioassay guided research approaches may facilitate the development of standardized extracts or purified compounds from Syzygium cumini that could serve as potential therapeutic alternatives with improved safety profiles for the management of lung cancer.
REFERENCES


Shrasti Singh, Dr. Nalini Kant Sahoo, Investigative Role of Syzygium Cumini Extract in Inhibiting A549 Lung Carcinoma Cell Proliferation, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 3, 2918-2940. https://doi.org/10.5281/zenodo.19200159
 10.5281/zenodo.19200159
10.5281/zenodo.19200159
