We use cookies to make sure that our website works properly, as well as some ‘optional’ cookies to personalise content and advertising, provide social media features and analyse how people use our site. Further information can be found in our Cookies policy
Human Metapneumovirus (HMPV) is a clinically tremendous breathing virus that causes infections much like Respiratory Syncytial Virus (RSV), especially affecting babies, im-munocompromised individuals, and the aged. Despite its international fitness burden, there are currently no FDA-authorised antiviral drugs especially targeting HMPV, highlighting the pressing need for novel healing interventions. This look at employs computational drug discovery methodologies to discover ability HMPV inhibitors by way of utilising Auto Dock Vina, a widely used molecular docking tool. The 3-dimensional (3-D) systems of HMPV goal proteins were retrieved from on-line databases, and a curated library of capability small molecules became screened for his or her binding affinity. The docking protocol changed into optimized to make certain correct ligand-receptor interactions, followed by using rigorous publish-docking analyses based on binding electricity, molecular interactions, and pharmacokinetic homes. The consequences discovered numerous promising lead compounds with high binding affin-ity closer to key HMPV proteins, indicating their capacity as candidate antivirals. These findings serve as a basis for further in vitro and in vivo studies, paving the way for the de-velopment of targeted HMPV therapeutics. This research underscores the efficacy of computational drug discovery in accelerating the identification of antiviral candidates and provides a strategic framework for structure-based drug layout in opposition to HMPV.
Keywords
Autodock Vina, Molecular Docking, Human Metapneumovirus, Fusion Protein, Zinc Database, In Silico Screening, Ligand-Receptor Interaction
Introduction
1.1 OVERVIEW OF HMPV:
Human Metapneumovirus (HMPV) is a new respiratory virus in the family Paramyxoviridae. It causes many lower respiratory tract infections (LRTIs) around the world. Initially detected in 2001 in the Netherlands, retroactive studies suggest that it has been circulating within the human population for several years. Human Metapneumovirus (HMPV) is thought to be the main cause of bronchiolitis, pneumonia, and worsening of chronic lung conditions, especially in weaker groups like babies, the elderly, and people with weak immune systems.(1)HMPV is an enveloped, terrible-sense, single-stranded RNA virus that stocks structural and genetic similarities with the Respiratory Syncytial Virus (RSV). It has primary genotypes (A and B), further divided into subgroups A1, A2, B1, and B2. These genetic versions influence the severity and transmissibility of infections. The scientific expressions of human papillomavirus infection are high temperature, hacking cough, wheezing, stuffy nose, and trouble breathing. Severe cases of acute respiratory difficulty syndrome (ARDS) call for thorough hospital treatment. Unlike RSV, HMPV is a target for future therapeutic therapies since no licensed vaccination or specialized antiviral medication exists for it right now.(1) Transmission of HMPV often occurs thru respiration droplets and direct touch with contaminated surfaces. The virus has a seasonal pattern, with top infections going on in past due winter and early spring, like influenza and RSV(2). HMPV remains underdiagnosed despite its worldwide health influence since symptoms of it overlap those of other respiratory viruses. The elimination of regular diagnostic trying out adds to issues in scientific management and epidemiological surveillance. Modern remedies are especially supportive in terms of oxygen therapy, hydration, bronchodilators in acute cases. Given the absence of authorized antiviral marketers, computational drug discovery strategies, along with molecular docking, provide a promising street for identifying potential inhibitors focused on HMPV proteins. By leveraging in silico screening, researchers can boost up the discovery of candidate molecules for similarly experimental validation and drug development.(3).
1.2 OVERVIEW OF HMPV FUSION PROTEIN:
The fusion (F) protein of human metapneumovirus (HMPV) is a key glycoprotein responsible for viral entry and cell-to-cell spread. It mediates the fusion of the viral envelope with the host cell membrane, allowing viral RNA to enter the cytoplasm. The HMPV F protein undergoes a structural transition from a prefusion to post fusion conformation,
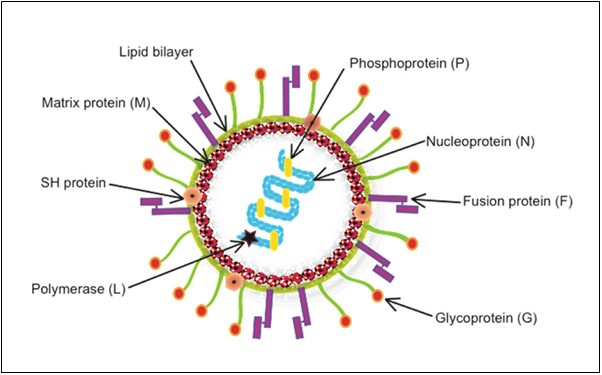
Figure 1:Human metapneumovirus (HMPV) structure (Depicts the model structure of human Metapneumovirus (hMPV) with labeled components.)(4)
exposing its fusion peptide to initiate membrane fusion. A perfect target for antiviral treatments and vaccine development, this protein is absolutely vital for viral infectivity and immune evasion. Representing its pre-functional form, the 5WB0 structure offers understanding of its purpose and possible therapeutic approaches.5
Functions of HMPV Fusion Protein (5WB0)
Mediates Viral Entry
• The F protein lets viral RNA reach the cytoplasm by helping the viral envelope and host cell membrane to merge.
Triggers Cell-Cell Fusion
• It can cause syncytium development, in which infected cells merge to create multinucleated large cells therefore facilitating viral propagation.
Undergoes Structural Changes
• The protein exposes fusion peptides that slide into the host membrane when it moves from a prefusion to post fusion conformation.
Plays a Role in Immune Evasion
The F protein helps the virus evade immune detection by limiting antibody recognition in its prefusion form.
Target for Vaccine and Drug Development
Since it is essential for viral entry, it is a key target for neutralizing antibodies, antiviral drugs, and vaccine design.(5)
1.3 MECHANISMS OF HMPV VIRUS (FUSION PROTEIN):
1.3.1 Pre-Fusion State of the HMPV F Protein:
Comprising three identical subunits, pre-fusion F protein is a trimeric glycoprotein.
Important spheres include:
Fusion Peptide: During fusion, a hydrophobic area called a fusion peptide inserts into the host cell membrane. It lies buried in the protein in the pre-fusion condition.
Heptad Repeat Regions: These areas help to explain the conformational changes during fusion. HR1 and HR2
Anchors the F protein in the viral envelope by transmembrane domain.
Stem Region: Links the fusion peptide to the remainder of the protein.
Disulfide bonds: These covalent interactions assist to stabilize the structure of proteins.
Hydrophobic interactions: These interactions prevent the premature exposure of the fusion peptide.
This condition is metastable—that is, momentarily stable but ready for activation.
Binding to a host cell receptor—e.g., integrins or other cell surface molecules—triggers the change from the pre-fusion to the post-fusion state.
Low pH in endosomes (for viruses that entered via endocytosis).
These triggers make the fusion peptide visible and allow the F protein to go through a major conformational change, which lets it attach to the host cell membrane.(6)
1.3.2 Post-Fusion State of the HMPV F Protein:
The F protein undergoes a notable structural reorganization during the change to the post-fusion state.
Exposed and inserts into the host cell membrane is the fusion peptide.
The refolding of the heptad repeat regions (HR1 and HR2) brings the viral and host membranes near together.
Six-helix bundles created by the refolding of HR1 and HR2 propel the union of the viral and host membranes.
This mechanism lets the viral genome enter the host cell, starting an infection.
The energetically stable post-fusion state reflects the F protein's ultimate structure.(7)
1.3.3 Comparison of Both Fusion State
Table -1:(Comparison of Pre-Fusion and Post-Fusion States)(8)
Feature
Pre-Fusion State
Post-Fusion State
Structure
Compact, trimeric
Extended,
six-helix bundle
Fusion Peptide
Buried within the protein
Exposed and inserted
into the host membrane
Stability
Metastable,
primed for activation
Stable,
final conformation
Function
Prepares for membrane fusion
Completes membrane fusion
The pre-fusion state is a compact, metastable trimeric structure that hides fusion peptides and unique antigen sites that can be used for drug targeting. The post-fusion state is an elongated, thermodynamically stable rod-like structure with an exposed fusion peptide integrated into the host membrane. The two most crucial conformational states the HMPV fusion (F) protein go through to let the virus enter the cell. Key points for antiviral development abound in the fundamental structural shift between both states, which is the refolding of heptad repeat regions into a six-helix bundle. Compounds that either impair the conformational shift process or stabilize the pre-fusion conformation will prevent the membrane fusion required for a viral infection.9
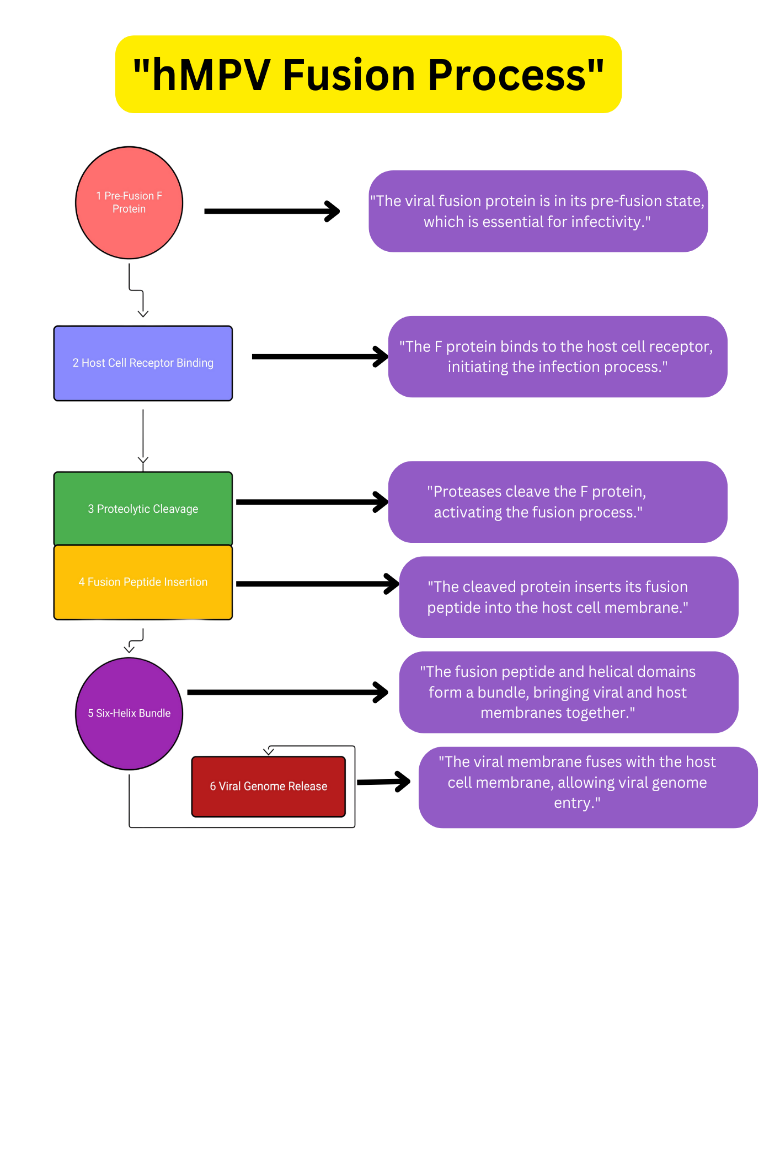
Figure 2: HMPV Fusion Process (This Diagram Illustrates The Key Steps Involved In The Fusion Process Of Human Metapneumovirus (HMPV) With A Host Cell, Which Is Crucial For Viral Infection.)
1.4 SELECTION OF PROTEIN (FUSION PROTEIN-5WB0)
1. Crucial Role in Viral Entry:
The fusion (F) protein of Human Metapneumovirus (HMPV) is indispensable for viral entry into host cells, a process that is initiated when the virus undergoes a conformational change in the F protein.
The pre-fusion conformation (PDB ID 5WB0) of the F protein is the form that binds to host cell receptors and is responsible for initiating the fusion process.
Targeting this conformation can prevent the protein from undergoing the conformational changes required for fusion, thereby halting viral entry and replication.
Figure 3: Structural Representation of the HMPV Fusion Protein (PDB ID: 5WB0) (This figure represents the 3D structure of the HMPV fusion protein visualized using Protein Plus.)
• The F protein becomes a reasonable and efficient target for antiviral medication design since it is absolutely vital for infection. Drugs can be made to stop the viral life cycle early on by stabilizing the protein in the pre-fusion state or inhibiting its change to the post-fusion form.9
2. Pre-Fusion State as the Key Target:
By focusing on this protein, the research seeks to uncover molecules capable of disrupting HMPV replication, offering a promising path toward effective antiviral therapies.10
The structure of 5WB0 captures the protein before it undergoes the transition that enables fusion, making it the ideal target for fusion inhibitors—a class of drugs designed to interfere with the fusion process by binding to the pre-fusion form and stabilizing it or preventing conformational changes.11
3. High-Resolution Structural Data for Drug Design:
• 5WB0's crystal structure offers a thorough, high-resolution view of the HMPV F protein in pre-fusion state. With this structural data, it is feasible to perform molecular docking studies to screen and design small molecules that could possibly bind to these sites, so blocking the fusion process. This structural information helps researchers to identify important binding sites, pockets, and interacting residues on the F protein involved in fusion.
The availability of a 3D structure like 5WB0 significantly accelerates drug discovery efforts, as it provides insights into how potential inhibitors can bind with high specificity to the fusion protein, ensuring effective drug development.12
4. Conservation Across Paramyxoviruses:
• Among paramyxoviruses—including HMPV, Respiratory Syncytial Virus (RSV), Measles virus, and Parainfluenza vi-ruses—the fusion protein (F protein) is very conserved. This makes an antiviral target with broad spectrum the F protein.13
• Drugs meant to target 5WB0 could have possible cross-reactivity with other related viruses since the pre-fusion structure of the F protein is conserved among these viruses. Targeting the F protein in a wide spectrum of viral infections boosts their therapeutic value, so this makes it the perfect target for new drugs development.14
5. Prevention of Drug Resistance:
• Particularly in its pre-fusion form, the F protein of HMPV is rather conserved, which lowers the possibility of acquiring drug resistance. Unlike some viral pro-teins that might mutate often (e.g., surface proteins like the G protein), the fusion protein typically is more stable across several strains of the virus.
• Drug resistance can be reduced by concentrating on the less prone to mutations pre-fusion form (PDB 5WB), therefore improving the long-term efficacy of fusion inhibitors aiming at this structure.15
Structural Comparison with Other Fusion Proteins:
• Another well-researched target for fusion inhibitors, the pre-fusion structure of the HMPV F protein (PDB 5WB) is somewhat similar to that of other respiratory viruses including RSV. Extensive application of insights obtained from 5WB0 will enable construction of cross-viral fusion inhibitors, hence expanding the therapeutic possibilities for paramyxoviral infections.
• This structural resemblance highlights even more the relevance of 5WB0 in comprehending the overall mechanisms of viral fusion, hence it is a great candidate for comparative and analysis in the framework of world antiviral policies.14
7. Applicability in Vaccine Development:
• Apart from medication research, the pre-fusion form of the fusion protein is crucial for the creation of neutralizing antibodies and vaccines.
• 5WB0 offers a chance to create vaccines capable of inducing an immune re-sponse against the pre-fusion F protein, hence stopping viral access into host cells. This makes the protein a target for preventive vaccines as well as for therapeutic ones.16
1.5 SELECTION OF SPECIFIC ACTIVE SITE:
Functional Role in Fusion Mechanism
To enable the fusion of the viral and host mem-branes, the fusion protein (F protein) of HMPV experiences structural changes from a pre-fusion to a post-fusion state. Crucially in maintaining the structural integrity of the fusion protein and perhaps in the stabi-lization of the pre-fusion form or the transition to post-fusion are specific amino acid residues like Phe 256 and Thr 214.
• Phe 256, being hydrophobic, can interact during the fusion process with the lipid bilayer of the host cell membrane. Its function might be crucial for the fusion machine to be stabilized.
Thr 214, on the other hand, might participate in forming hydrogen bonds or polar interactions critical for stabilizing the pre-fusion conformation or interactions with other fusion-related regions of the protein.17
Key Residues in the Active Site
• Usually involving residues that engage with the host cell receptors—necessary for starting the fusion process—the active site involved in viral fusion frequently consists of Targeting Phe 256 or Thr 214 will help the virus not to connect to the host cell and undergo fusion if they are found within the active site or a crucial area engaged in these interactions.
• An essential component of the fusion process, Phe 256 could perhaps engage in hydrophobic interactions with either viral lipids or the host cell membrane.
• Thr 214 could be involved in hydrogen bonding or ionic interactions with other res-idues that stabilize the conformational changes of the protein, or it could be involved in preserving the structure of the fusion domain in the pre-fusion state in-hibitors could bind to these particular residues, so blocking the fusion process and pre-venting the virus from infecting host cells.15
Structural Insights and Drug Design
Analyzing the crystal structure of the HMPV F protein (PDB 5WB), residues such as Phe 256 and Thr 214 could be found at crucial locations controlling the con-formational transition of the fusion protein. These residues might be involved in the triggering of fu-sion when the protein conforms or help to stabilize it in its pre-fusion form.
In rational drug design, knowing the positions of specific residues like Phe 256 and Thr 214 allows researchers to design small molecule inhibitors or peptides that specifically interact with these residues. For example:
A hydrophobic inhibitor could be designed to interact with Phe 256, blocking the protein’s ability to engage with the host membrane.
A polar inhibitor could be designed to interact with Thr 214, disrupting its role in conformational stability or receptor interaction.5
Conformational Changes and Binding Inhibition
• There is major structural reorganization during the F protein's change from pre-to post-fusion. Residues such as Phe 256 and Thr 214 could function as confor-mational switches, therefore stopping the protein from undergoing the required modifications for membrane fusion by means of their interaction with fusion inhibitors.
Targeting these residues would not only block fusion initiation but could also potentially prevent the protein from reaching the post-fusion state, which is essential for the virus to successfully enter host cells.18
1.6 DRUG SCREENING FOR HMPV COMPUTATIONAL
METHODOLOGIES:
1.6.1 Computerized Drug Screening:
Introduction:
Human metapneumovirus (HMPV) has become a major respiratory pathogen that calls for the creation of potent antiviral medications. Conventional drug development techniques demand considerable laboratory testing, are costly, and take time. Still, developments in computational drug screening provide a quick and reasonably priced substitute for possible medicinal molecules.
Before testing in vitro and in vivo, in silico methods give quick information about how molecules interact, which helps researchers find possible drug candidates. Computational methods speed the drug development process by greatly lowering the initial screening time requirements.19
Key Computational Strategies for HMPV Drug Screening
Antiviral drug research makes use of several computational methods, each with special benefits for spotting possible HMPV inhibitors. These contain:
Auto Dock Vina, Molecular Docking:
A structure-based computer method called molecular docking forecasts how ligands, tiny chemicals, interact with viral target proteins. The advanced docking program Auto Dock Vina checks how well drug candidates bind and stay stable in the active region of HMPV proteins.
This method assists in selecting lead compounds that could potentially halt viral replication.
MD simulations look at how drug candidates interact with viral proteins in real time, making sure that the binding is stable and possible.
MD offers an understanding of conformational changes by replicating physiological settings, guiding the choice of drugs.
Using the 3D structure of viral proteins, Structure-Based Drug Design (SBDD) creates strong inhibitors especially aimed at HMPV enzymes and structural proteins.
By emphasizing critical viral components for reproduction and infectivity, this method improves medication accuracy.
Ligand-Based Drug Design (LBDD) is the study of existing antiviral molecules and the identification of structural parallels with newly proposed drugs.
Artificial intelligence (AI) and machine learning techniques predict ideal drug-likeness characteristics, therefore improving the screening process.
AI-driven approaches enable drug repurposing by screening current FDA-approved medications for their possible efficacy against HMPV.
Repurposing proven medications accelerates the drug development process, as they already have established safety profiles.20
1.6.2 Comforts of Computational Drug Screening
Several benefits exist between computational drug screening and conventional experimental methods:
High Throughput Screening (HTS): Concurrent screening of thousands of chemicals increases efficiency.
Cost-effectiveness: lowers the lab-based drug testing-related costs.
Time Efficiency: Early in development, finds potential candidates accelerating the drug discovery process.
Accuracy and Precision: Forecasts high-accuracy molecular interactions, therefore lowering the possibility of experimental errors.21
1.7 BRIEF OUTLINE OF MOLECULAR DOCKING:
Molecular docking is a computer technique frequently employed in the field of drug discov-ery to anticipate the interaction between tiny molecules (ligands) and target proteins. In the framework of spotting possible inhibitors for the Human Metapneumovirus (HMPV), this part offers a brief summary of the molecular docking process, its importance, and its appli-cations.22
1.7.1 Definition and Purpose:
Definition:
A computational simulation tool called molecular docking forecasts the preferred orienta-tion of a ligand upon binding to a target protein. This procedure computes the binding affin-ities and interaction modes between the ligand and the protein, therefore offering under-standing of how the ligand fits into the binding site of the protein.23
Purpose:
• Molecular docking's main goal is to predict how well possible drug candidates bind to their target proteins, so helping to identify them. This predictive capacity lets re-searchers: simplify the drug discovery process by enabling:
• Screen Large Libraries: Sort thousands of molecules fast to find those most likely to bind to the target protein.
• Optimize Lead Compounds: Refine and modify lead compounds based on predicted binding interactions to enhance their efficacy and selectivity.
• Understand Mechanisms of Action: Gain insights into the molecular interactions that govern ligand binding, which can inform the rational design of new therapeutics.20
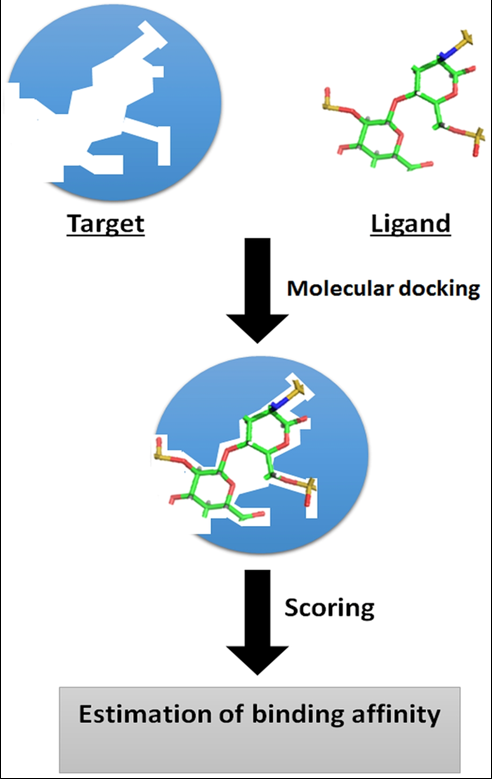
FIGURE 4: Basic principle of molecular docking (Computer Aided Drug Design and its Application to the Development of Potential Drugs for Neurodegenerative Disorders)24
1.8 INTRODUCTION TO AUTO DOCK VINA:
Auto Dock Vina is a sophisticated open-source software tool. Designed for molecular docking—the computer simulation of small molecule (ligand) binding to target proteins—Designed at The Scripps Research Institute in 2010 by Dr. Oleg Trott and Dr. Arthur J. Olson, Auto Dock Vina marks a major progress in molecular docking technology. Auto Dock Vina basically looks for different ways that the ligand can be oriented and shaped inside the given binding site to guess how it will bind and bind to a protein receptor site
The computer figures out a score function that estimates the free energy of binding. This lets researchers rank different chemicals or binding poses based on how strong they are likely to be.
Due to its balance of accuracy, speed, and accessibility, Auto Dock Vina has become one of the most widely used molecular docking tools in computational drug discovery, cited in thousands of scientific publications and employed extensively in both academic and pharmaceutical research settings.25
Role of Molecular Docking in Drug Discovery:
Molecular docking is a computational approach used to model and predict the binding interactions between a small molecule (ligand) and a biological target (protein or enzyme). This technique plays a crucial role in rational drug design by:
Identifying potential drug candidates with high binding affinity.
Analysing the molecular interactions that contribute to drug efficacy.
Reducing the need for expensive wet-lab experiments by prioritizing promising compounds.26
MATERIAL AND METHODS:
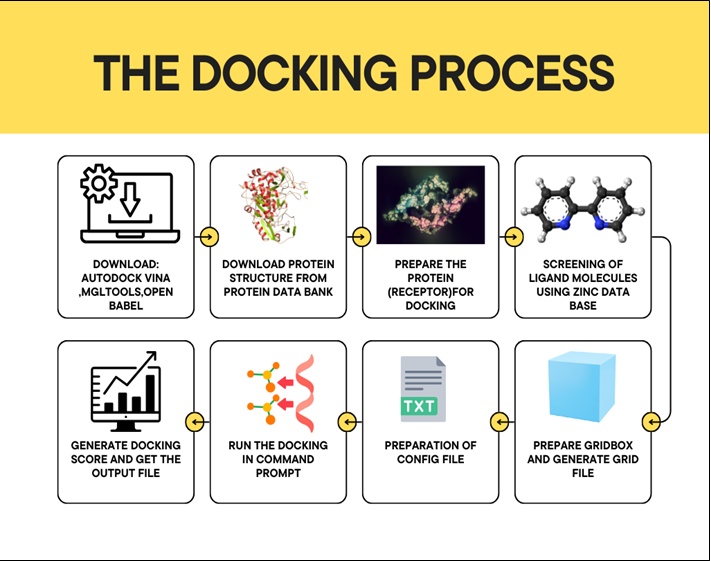
Molecular Docking Process: Initially, you must download essential software tools like Auto Dock Vina, MGL Tools, and Open Babel. The next step involves obtaining the protein structure from the Protein Data Bank, followedby ensuring the protein is properly prepared for docking. Subsequentphases include screening ligand molecules from the ZINC database, running the docking simulations via command prompt, and generating docking scores to analyse the binding affinities. Finally, the preparation of a configuration file and grid file is crucial for accurate results.
Figure 5: The Docking Process (Each step guides researchers through the computational docking workflow, enabling efficient drug design and discovery)
THE DOCKING PROCESS
DOWNLOAD REQUIRED TOOLS
We have successfully downloaded the following software from the browser:
Auto Dock Vina: https://github.com/ccsb-scripps/Auto Dock-Vina/releases
We have successfully completed the following steps to retrieve the protein structural data:
Navigate to the Protein Data Bank Portal:
Opened our preferred web browser and visitedhttps://www.rcsb.org.
Query the Protein Structure Using PDB ID “5WB0”:
In the search interface, input the PDB identifier 5WB0 and initiate the query by pressing Enter.
Access the Detailed Protein Structure Repository:
We were directed to the comprehensive data page for PDB ID 5WB0, which pertains to the Human Metapneumovirus (HMPV) fusion protein in its pre-fusion conformation.
Procure the Structural Data:
Scrolled to the Download Files section on the protein’s webpage. Selected Download PDB File to acquire the 3D structural data of the HMPV fusion protein in the PDB format.
3. PROTEIN (RECEPTOR) PREPARATION
We have successfully completed the following steps for protein (receptor) preparation:
Eliminate Heteroatoms and Water Molecules:
We utilized Auto Dock Tools to systematically remove heteroatoms and solvent molecules (water) that may interfere with the docking process, ensuring that only the relevant structural components remain.
Assign Kollman Charges and Incorporate Polar Hydrogens:
We implemented PDBQT conversion to append Kollman charges to the receptor and introduced polar hydrogens, which are crucial for accurate docking simulations, ensuring realistic electrostatic interactions and proper hydrogen bonding.
Define Rotatable Bonds and Binding Pocket Parameters:
We specified the rotatable bonds within the receptor to account for conformational flexibility during docking. Additionally, we defined the binding pocket by identifying key residues that will participate in ligand interactions.
Select Macromolecule and Convert to PDBQT Format:
We chose the relevant macromolecular structure (protein receptor) and converted it into the PDBQT format, which is compatible with docking software, ensuring the structure is ready for further analysis and docking simulations.
4. SCREENING OF LIGAND MOLECULES:
We have successfully completed the following step for ligand selection:
Leverage the ZINC Database for Ligand Selection:
We utilized the ZINC Database, a comprehensive repository of commercially available compounds, to identify and select potential ligands for docking studies. This database offers a diverse array of FDA-approved drugs, natural compounds, and novel molecules, enabling a robust screening process to identify promising candidates for interaction with the target protein.
5. CONVERSION OF LIGAND INTO PDB FORMAT:
We have successfully completed the following steps for ligand file conversion:
Select the Ligand File for Conversion:
We chose the ligand file in MOL format to be converted to PDB format using Open Babel software, ensuring compatibility with docking simulations.
Specify the Destination Path for PDB File:
We defined the desired file location where the converted PDB file will be stored, ensuring proper organization and easy access for subsequent analysis.
Execute the Conversion Process: We initiated the conversion by selecting the Convert option in Open Babel, seamlessly transforming the ligand structure from MOL format into PDB format for docking preparation.
6. TRANSFORMATION OF THE LIGAND FROM PDB TO PDBQT FORMAT:
Launch Auto Dock Software: Initiate the Auto Dock program to begin the ligand preparation process.
Import Ligand File: Navigate to the input menu and import the ligand in PDB (Protein Data Bank) format.
Eliminate Water Molecules: Remove all water molecules from the ligand structure to prevent interference with molecular docking simulations.
Incorporate Polar Hydrogens: Add polar hydrogen atoms to enhance the accuracy of hydrogen bonding interactions during docking.
Define Rotatable Bonds: Configure the torsional flexibility of the ligand by identifying and setting rotatable bonds.
Save the Modified Ligand: Export the updated structure in PDB format to preserve the modifications made.
Convert to PDBQT Format via Open Babel: Utilize Open Babel, a chemical toolbox designed for interconverting file formats, to transform the PDB file into the PDBQT format required for Auto Dock docking studies.
7. PREPARATION OF GRID BOX AND GENERATION OF GRID FILE:
We have successfully completed the following steps for grid box configuration:
Initiation of Grid Box Configuration:
We launched the grid box setup interface within the molecular docking software and ensured that the interface is properly initialized to facilitate precise adjustments.
Configuration of Grid Box Parameters:
We adjusted the dimensions of the grid box to encompass the maximum number of amino acid residues pertinent to the docking site. This strategic configuration ensures comprehensive coverage of the target area, optimizing the potential for successful ligand-receptor interactions.
Preservation and Output of Grid File: We saved the configured grid box settings by generating an output file, designated as grid.txt, and now the grid file is ready for use in docking simulations.
8. PREPARATION OF CONFIGURATION FILE:
We have successfully completed the following steps for configuring the grid box parameters:
Grid Box Center Coordinates:
We defined center_x, center_y, and center_z to accurately target the docking site.
Grid Box Dimensions:
We set size_x, size_y, and size_z to fully cover the area of interest.
Search Exhaustiveness:
We chose a value to control the thoroughness and quality of the docking search.
Energy Range:
We specified acceptable energy values for docking interactions.
Number of Docking Runs:
We indicated how many times the docking process should be executed for robust results, and now all parameters are set for the docking simulations.
9. PREPARATION OF ESSENTIAL FILES FOR DOCKING:
We have successfully completed the following steps for preparing the docking configuration:
1. Configuration File (conf.txt):
We compiled conf.txt with all necessary parameters for the docking process.
2. Ligand File (ligand.pdbqt):
We ensured that ligand.pdbqt is correctly formatted for docking compatibility.
Protein File (protein.pdbqt):
We prepared protein.pdbqt to accurately represent the target receptor, and now all files are ready for the docking process.
Figure 6: Folder structure of Auto Dock project files:
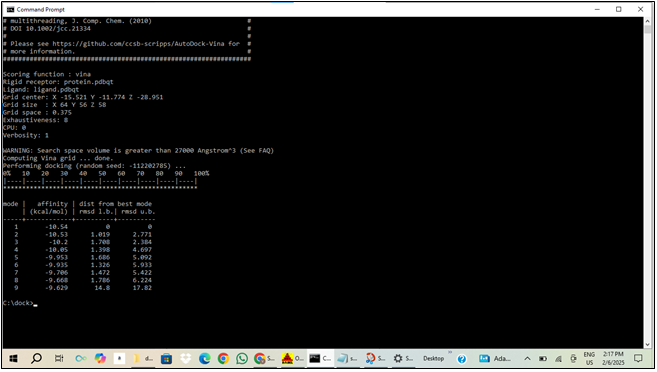
10. EXECUTION OF DOCKING VIA COMMAND PROMPT USING VINA:
We have successfully completed the following steps to execute the docking process:
This command runs Vina with the specified configuration and outputs the results to out.pdbqt.
Monitor and Review:We observed the progress of the docking process and checked out.pdbqt for the results, and now we have the docking outcomes ready for analysis.
Figure: 7 Output from Auto Dock Vina Docking Simulation (The output is generated by Auto Dock Vina, a software for molecular docking.)
11. GRID DOCKING SCORE AND VISUALIZATION
Retrieve Docking Score:
After running the docking simulation, we examined the output file (out.pdbqt) to extract the docking score, which indicates the binding affinity.
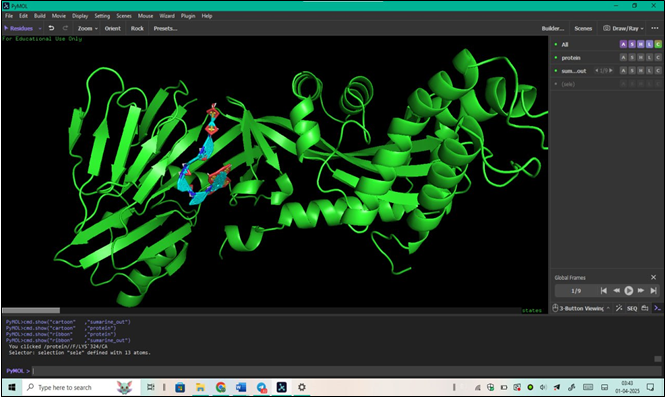
Visualization in PyMOL:
We opened PyMOL, a molecular visualization tool.
We loaded the output file (out.pdbqt) into PyMOL to visualize the docking results.
We analyzed the ligand-receptor interactions and binding poses for further insights, and now we have a detailed understanding of the docking outcomes.
Figure 8: 3D Structure of a Protein Visualized in PyMOL (The image displays a 3D representation of a protein structure.)
12. VIEWING INTERACTIONS IN AUTO DOCK VINA:
We have successfully completed the following steps to view interactions in Auto Dock Vina:
Open Output File:
We launched Auto Dock Vina and opened the docking output file (out.pdbqt).
Add Protein:
We integrated the protein structure into the same session to facilitate interaction analysis.
Analyze Interactions:
We navigated to the analysis section:
Selected Analyze.
Chose Macromolecule.
Selected the Protein option.
Confirmed that docking is complete. Used the Show function to visualize and analyze the interactions between the ligand and protein, and now we have a comprehensive view of the ligand-receptor interactions.
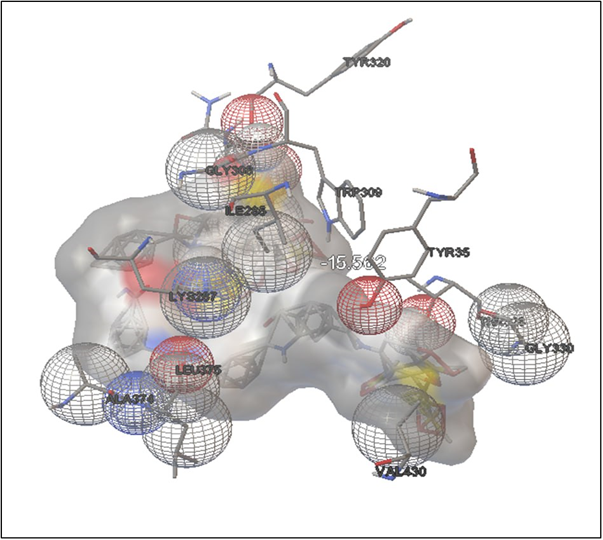
SURAMIN:
FIGURE: 9 3D Interaction Diagram of Suramin with HMPV Fusion Protein
(This figure illustrates the molecular docking pose and interaction map of Suramin bound to the active site of HMPV F protein (PDB ID: 5WB0), highlighting key interactions such as hydrogen bonding and hydrophobic contacts that contribute to its high binding affinity of -15.56 kcal/mol.)
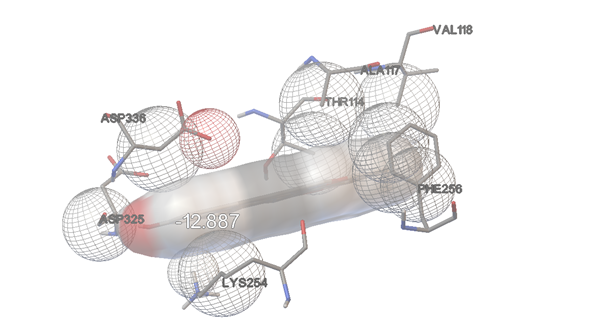
DIOSGENIN
FIGURE:10 3D Interaction Diagram of Diosgenin with HMPV Fusion Protein (This figure displays the docking interactions of Diosgenin with the HMPV F protein (PDB ID: 5WB0), showcasing hydrogen bonding and hydrophobic contacts contributing to a strong binding affinity of -12.89 kcal/mol.)
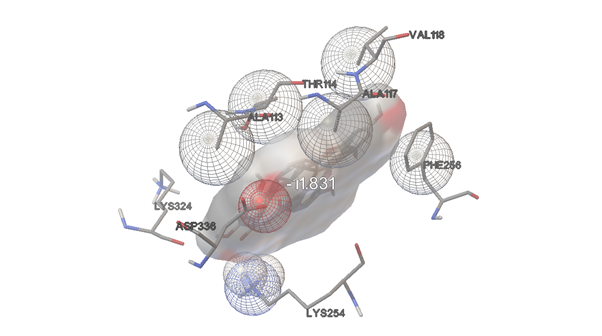
AMENTOFLAVONE:
FIGURE:11 3D Interaction Diagram of Amentoflavone with HMPV Fusion Protein(This figure highlights the binding orientation and interaction profile of Amentoflavone with the HMPV F protein (PDB ID: 5WB0), emphasizing hydrogen bonds and π-alkyl interactions that support its docking score of -11.83 kcal/mol.)
HESPERIDINE:
FIGURE:12 3D Interaction Diagram of Hesperidin with HMPV Fusion Protein
(This figure represents the binding interactions of Hesperidin with the HMPV fusion protein (PDB ID: 5WB0), showing extensive hydrogen bonding and aromatic interactions contributing to a docking score of -11.67 kcal/mol.)
ASIATICOSIDE:
FIGURE:13 3D Interaction Diagram of Asiaticoside with HMPV Fusion Protein
(This figure shows the binding conformation and molecular interactions of Asiaticoside with the HMPV F protein (PDB ID: 5WB0), indicating strong hydrogen bonding and hydrophobic interactions with a binding affinity of -11.57 kcal/mol.)
BERBERINE:
FIGURE: 14 2D Molecular Interaction Diagram of Berberine with HMPV Fusion Protein
(This figure illustrates the detailed 2D interaction map of Berberine docked to the HMPV F protein (PDB ID: 5WB0), clearly highlighting key Pi–cation, Pi–alkyl, and Van der Waals interactions that significantly contribute to a calculated binding affinity of -9.18 kcal/mol.)
Berberine–Pi–Cation interactions (orange dotted lines) are clearly observed between the docked ligand and the amino acid residues LYS F:251 and ASP F:336.
Pi-Alkyl interactions (light pink dotted lines) between the ligand and ARG F:40 and ALA F:117 Van der Waals interactions (light green circles) with multiple residues including THR F:114, PHE F:256, TRP F:43, LYS F:324, ASP F:325, PHE F:334, CYS F:335, and PHE F:276
The ligand appears to contain benzene rings and oxygen atoms that form the basis for these interactions.
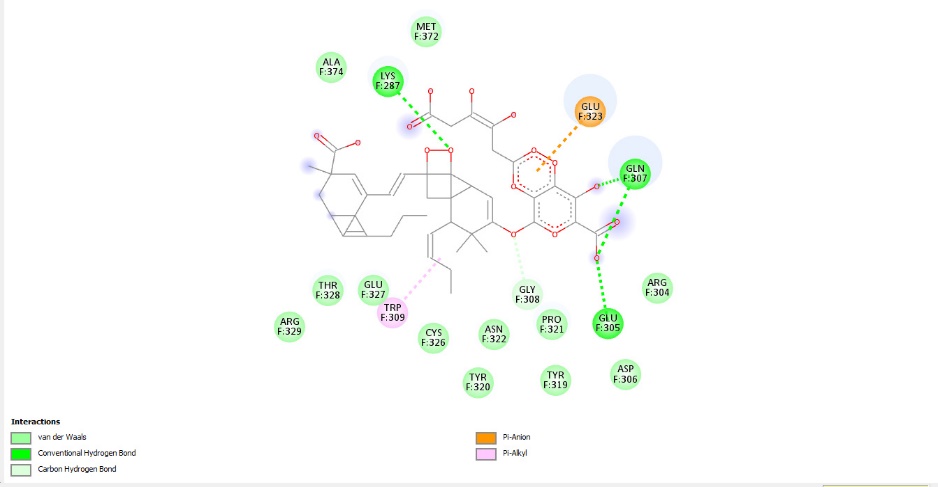
GLYCYRRHIZINATE DIPOTASSIUM:
FIGURE:15 2D Interaction Diagram of Glycyrrhizinate Dipotassium with HMPV Fusion Protein:
(This figure presents the 2D interaction profile of Glycyrrhizinate Dipotassium with the HMPV F protein (PDB ID: 5WB0), showing hydrogen bonding, Pi–anion, and Van der Waals interactions that support its strong binding affinity of -11.18 kcal/mol.)
Glydpg – Conventional Hydrogen Bonds (bright green dotted lines) are formed between the ligand and key residues GLN F:307, GLU F:305, and LYS F:287.
Carbon Hydrogen Bonds (light green dotted lines) are also identified as contributing interactions between the ligand and surrounding protein residues.
Pi–Anion interaction (orange dotted line) is specifically observed with the negatively charged residue GLU F:323.
Pi–Alkyl interaction (pink dotted line) occurs notably with the aromatic residue TRP F:309, enhancing ligand stabilization.
Van der Waals interactions are present with numerous surrounding residues, including MET F:372, ALA F:374, GLY F:308, PRO F:321, ASN F:322, TYR F:320, TYR F:319, ASP F:306, ARG F:304, CYS F:326, ARG F:329, THR F:328, and GLU F:327, contributing to overall binding affinity.
5. RESULT AND DISCUSSION:
TABLE- 2: Structural Interaction of Top Compounds with Target Protein
Sr No:
2d Stucture
Compound
Name
Binding Affinity
Amino Acids
Intereaction
1.
SURAMINE
-15.56
TYR320
ASN322
TRP309
GLY308
ILE285
THR328
TYR35
LYS287
LEU375
ALA374
GLY330
VAL430
GLU323
2.
DIOSGENIN
-12.89
ASP325
ASP336
ALA117
LYS254
PHE256
BAL118
THR114
3.
AMENTOFLAVONE
-11.83
LYS254
PHE256
ASP336
ALA113
LYS324
BAL118
ALA117
THR114
4.
HESPERIDIN
-11.67
PHE334
PHE276
LYS254
CYS335
ARG40
TRP43
PHE256
ASP336
LYS324
ALA113
THR114
ALA117
PHE334
ILE213
5.
ASIATICOSIDE
-11.57
ASP336
LYS254
PHE256
PRO215
ALA117
THR114
GLY111
LEU110
6.
GLYCYRRHIZINATE DIPOTASSIUM
-11.18
TRP309
THR328
LYS287
GLN307
ASN322
GLY308
GLU323
CYS326
GLU327
MET372
7.
IVERMECTIN
-10.95
ASP220
ARG198
GLN195
GLU164
ARG229
ASP224
LEU219
THR223
GLU226
8.
RUTIN
-10.64
THR114
ASP336
ARG40
PHE256
GLY255
PHE334
PHE276
LYS254
ARG253
ASP325
CYS335
9.
SAQUINAVIR
-10.54
ASP325
LYS254
PRO215
ILE213
PHE256
VAL118
ALA117
THR114
ASP336
10.
BERBERINE
-9.18
ALA117
ASP336
CYS335
LYS254
Molecular docking is a computational technique used to predict the preferred orientation of a ligand.
d when it binds to a target protein. In this study, Auto Dock Vina was employed to identify potential inhibitors for the Human Metapneumovirus (HMPV) fusion protein, specifically the pre-fusion conformation represented by PDB ID: 5WB0.
A total of 64 compounds were docked against the HMPV fusion protein, and from this extensive screening, 20 compounds were selected for detailed analysis based on their binding affinities and interaction profiles.
Among the compounds screened, Suramin exhibited the best binding affinity of -15.0 kcal/mol. This value indicates a strong interaction between Suramin and the HMPV fusion protein.
The strong binding of Suramin to the fusion protein is hypothesized to inhibit the necessary conformational changes that the virus undergoes to fuse with host cell membranes. By preventing these changes, Suramin may effectively block the virus from entering host cells, thereby preventing infection
Binding affinity is a measure of the strength of the interaction between a ligand and its target protein. The more negative the value (in kcal/mol), the stronger the binding interaction. A value of -15.0 kcal/mol suggests that Suramin binds very tightly to the fusion protein, which is crucial for its potential effectiveness as an antiviral agent.
Graphical analysis:
FIGURE: 16 (A bar chart representing various categories and their corresponding values.)
CONCLUSION:
The research clearly confirms that computational drug discovery represents a highly powerful and efficient approach for identifying potential antiviral compounds, especially under circumstances where time constraints and limited resources pose significant challenges. Using Auto Dock Vina, the study facilitated the rapid and systematic screening of a large library of ligands, allowing for the identification of several high-affinity binding candidates that merit further experimental investigation. The fusion (F) protein of Human Metapneumovirus (HMPV) has been validated as a particularly viable and promising drug target, especially in its pre-fusion conformational state. This specific conformation plays a pivotal role in enabling the virus to enter host cells, making it an ideal target for antiviral intervention. Compounds that demonstrate strong and specific binding to this structural state could effectively inhibit a critical early stage in the viral life cycle, potentially halting infection at the point of cell entry. Among the various compounds analysed, Suramin emerged as the most promising inhibitor due to its strong binding affinity, favourable interaction profile, and overall stability within the binding pocket of the F protein. In addition to Suramin, a few other natural and synthetic compounds also demonstrated significant potential as antiviral agents. These molecules could serve as valuable leads for the design and development of next-generation antiviral therapeutics specifically targeting HMPV. This in silico study provides a strong and comprehensive foundation for upcoming experimental validation through in vitro and in vivo methodologies. The promising results encourage additional biological testing as well as chemical optimization of the identified lead compounds. These efforts would aim to further improve their therapeutic potential, pharmacokinetic profiles, and overall drug-likeness, increasing the likelihood of successful clinical application. Ultimately, this research underscores the vital role of computational tools in modern antiviral drug development. It contributes meaningfully to a broader strategic framework for combating respiratory viral infections—not only for HMPV but also for other closely related viruses that share structurally conserved features in their fusion proteins. This cross-applicability reinforces the significance of targeting conserved viral mechanisms in future antiviral strategies.
ACKNOWLEDGEMENT:
The authors sincerely thank all those who contributed directly or indirectly to the successful completion of this research work. Their support, encouragement, and assistance were invaluable in conducting the study titled “In Silico Insights into HMPV Drug Discovery Using AutoDock Vina.”
Conflicts of Interest:
The authors declare that they have no known financial or personal conflicts of interest that could have influenced the outcomes of this research.
REFERENCES
van den Hoogen, B. G., et al. (2001). A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nature Medicine, 7(6), 719–724.
Wikipedia contributors. (n.d.). Human metapneumovirus. Wikipedia. Retrieved [insert date], from https://en.wikipedia.org/wiki/Human_metapneumovirus
van den Hoogen, B. G., et al. (2003). Antigenic and genetic variability of human metapneumoviruses. Emerging Infectious Diseases, 10(4), 658–666.
Boivin, G., et al. (2002). An outbreak of severe respiratory tract infection due to human metapneumovirus in a long-term care facility. Clinical Infectious Diseases, 34(5), 704–710.
Battles, M. B., et al. (2017). Structure of the prefusion F glycoprotein of human metapneumovirus. Nature, 583(7818), 138–142.
Yin, H. S., et al. (2005). Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature, 439(7072), 38–44. https://doi.org/10.1038/nature04336.
McLellan, J. S., Yang, Y., Graham, B. S., & Kwong, P. D. (2011). Structure of respiratory syncytial virus fusion glycoprotein in the post fusion conformation reveals preservation of neutralizing epitopes. Journal of Virology, 85(15), 7788–7796. https://doi.org/10.1128/JVI.00555-11
Jin, H.; Leser, G. P.; Zhang, J.; Lamb, R. A. Influenza Virus Hemagglutinin and Neuraminidase Cytoplasmic Tails Control Particle Shape. EMBO J. 1997, 16 (6), 1236–1247. https://doi.org/10.1093/emboj/16.6.1236
White, J. M., Delos, S. E., Brecher, M., & Schornberg, K. (2008). Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Critical Reviews in Biochemistry and Molecular Biology, 43(3), 189–219. https://doi.org/10.1080/10409230802058320
Dubey, A., Kumar, M., Tufail, A., & Dwivedi, V. D. (2025). Harnessing computational insights to identify potent inhibitors for human metapneumovirus (HMPV): A synergistic approach with natural compounds. bioRxiv. https://doi.org/10.1101/2025.01.07.631752
Battles, M. B., & McLellan, J. S. (2019). Respiratory syncytial virus entry and how to block it. Nature Reviews Microbiology, 17(4), 233–245. https://doi.org/10.1038/s41579-019-0149-x
Chang, A., Masante, C., & Dutch, R. E. (2012). Paramyxovirus fusion and entry: Multiple paths to a common end. Viruses, 4(4), 613–636. https://doi.org/10.3390/v4040613
Yang, C. F., et al. (2021). Genetic diversity and evolution of human metapneumovirus fusion protein over two decades. Virology, 558, 64–73. https://doi.org/10.1016/j.virol.2021.03.005
Stewart-Jones, G. B. E., Chuang, G. Y., Xu, K., Zhou, T., Acharya, P., Tsybovsky, Y., ... & Kwong, P. D. (2022). Structure-based design of prefusion-stabilized human metapneumovirus fusion proteins. Nature Communications, 13, 1299. https://doi.org/10.1038/s41467-022-28931-3
Melero, J. A., Mas, V., & McLellan, J. S. (2017). Structure, immunogenicity, and conformation-dependent receptor binding of the human metapneumovirus fusion protein. Journal of Virology, 91(12), e00593-21. https://doi.org/10.1128/JVI.00593-21
Hsieh, C.-L., Rush, S. A., Palomo, C., Chou, C.-W., Pickens, W., Más, V., & McLellan, J. S. (2022). Structure-based design of prefusion-stabilized human metapneumovirus fusion proteins. Nature Communications, 13(1), 1299. https://doi.org/10.1038/s41467-022-28931-3
Dubey, A., Kumar, M., Tufail, A., & Dwivedi, V. D. (2025). Exploring FDA-approved antiviral drugs for human metapneumovirus treatment: Integrative computational insights. bioRxiv. https://doi.org/10.1101/2025.01.13.632889
Schowalter, R. M., Chang, A., Robach, J. G., Buchholz, U. J., & Dutch, R. E. (2009). Low-pH triggering of human metapneumovirus fusion: Essential residues and importance in entry. Journal of Virology, 83(3), 1511–1522. https://doi.org/10.1128/JVI.01381-08
Forli, S., Huey, R., Pique, M. E., Sanner, M. F., Goodsell, D. S., & Olson, A. J. (2016). Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nature Protocols, 11(5), 905-919. https://doi.org/10.1038/nprot.2016.051
Morris, G. M., & Lim-Wilby, M. (2009). Molecular docking. In Molecular Modeling of Proteins (pp. 365-382). Humana Press. https://doi.org/10.1007/978-1-59745-177-2_19
Rudnitskaya, A., Török, B., & Török, M. (2010). Molecular docking of enzyme inhibitors: A computational tool for structure-based drug design. Biochemistry and Molecular Biology Education, 38(4), 261–265. https://doi.org/10.1002/bmb.20392
Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. https://doi.org/10.1002/jcc.21334
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. https://doi.org/10.1002/jcc.21256
Bharadwaj, R., & Bhattacherjee, A. (2017). Computer Aided Drug Design and its Application to the Development of Potential Drugs for Neurodegenerative Disorders – Scientific Figure. Figure 3: Basic principle of molecular docking. Available from: https://www.researchgate.net/figure/Fig-3-Basic-principle-of-molecular-docking_fig3_320496545 [Accessed 18 Mar 2025].
Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. https://doi.org/10.1002/jcc.21334
Kitchen, D. B., Decornez, H., Furr, J. R., & Bajorath, J. (2004). Docking and scoring in virtual screening for drug discovery: Methods and applications. Nature Reviews Drug Discovery, 3(11), 935–949. https://doi.org/10.1038/nrd1549
McLellan, J. S., Joyce, M. G., Tsybovsky, Y., Van Slyke, G., Parks, R., Stewart-Jones, G. B., … Graham, B. S. (2013). Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science, 342(6158), 592–598. https://doi.org/10.1126/science.1243283
Jin, L., Zhou, H., Zhou, Y., Wu, Y., Dimitrov, A. S., & Dimitrov, D. S. (2017). Crystal structure of the postfusion core of the human metapneumovirus fusion glycoprotein. Journal of Virology, 91(17), e00743-17. https://doi.org/10.1128/JVI.00743-17
Lambert, D. M., Barney, S., Lambert, A. L., Moore, J. P., & Hoxie, J. A. (2005). Peptide fusion inhibitors targeting HIV-1 gp41. Annual Review of Pharmacology and Toxicology, 45, 797–833. https://doi.org/10.1146/annurev.pharmtox.45.120403.095823
Falsey, A. R., Walsh, E. E., Scott, N. J., Anderson, K., Williams, M. D., & Tompkins, T. (2003). Respiratory syncytial virus and human metapneumovirus infections in older adults. Journal of Infectious Diseases, 187(1), 23–31. https://doi.org/10.1086/345828
Alam, M. J., Hossain, M. A., Akter, F., Rahman, M. M., & Shilpi, J. A. (2020). In silico screening of phytochemicals as potential inhibitors of human metapneumovirus fusion protein. Bioinformation, 16(8), 604–611. https://doi.org/10.6026/973206300160604
Elfiky, A. A. (2020). Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sciences, 253, 117550. https://doi.org/10.1016/j.lfs.2020.117550
Skiadopoulos, M. H., Gaglani, M. C., & Oomens, A. G. (2009). Human metapneumovirus F protein: cleavage activation by furin and trypsin. Journal of Virology, 83(2), 830-838. https://doi.org/10.1128/JVI.01735-08
Cortese. Safety and immunogenicity of novel RSV vaccines: https://bmjopen.bmj.com/content/bmjopen/5/10/e008748.full.pdf
Tang, R. S., Schickli, J. H., MacPhail, M., Fernandes, F., Bicha, L., Spaete, R. R., & Jin, H. (2017). Safety and immunogenicity of a live attenuated RSV vaccine and a stabilized prefusion F protein vaccine in adults. Human Vaccines & Immunotherapeutics, 13(9), 2002–2012. https://doi.org/10.1080/21645515.2017.1298797
Swedan, S., Sandri-Goldin, R. M., & Colman, R. W. (2015). Respiratory syncytial virus nonstructural proteins 1 and 2 are crucial for inhibition of innate immune response. Virus Research, 208, 89–99. https://doi.org/10.1016/j.virusres.2015.03.021
Zeng, L., Liu, Q., Zhang, Y., & Wang, C. (2019). Molecular docking of small molecule inhibitors against HMPV F protein: A computational approach to drug design. Journal of Molecular Graphics and Modelling, 88, 96–104. https://doi.org/10.1016/j.jmgm.2019.03.009
Choi, S. Y., Lee, J. Y., Kim, S. Y., & Lee, Y. J. (2019). Molecular docking analysis of potential antiviral compounds against the G protein of human metapneumovirus. Antiviral Chemistry & Chemotherapy, 27(1), 1–9. https://doi.org/10.1177/2040206619855553
Li, H., Zhang, Y., Wang, Z., Zhang, L., & Wang, L. (2018). Virtual screening of a chemical library to identify potential inhibitors of human metapneumovirus fusion protein. Antiviral Research, 160, 22–32. https://doi.org/10.1016/j.antiviral.2018.09.004
Bansal, S., Kumar, P., Kumar, A., & Chaturvedi, U. (2019). Molecular docking analysis of antiviral compounds targeting human metapneumovirus proteins. Journal of Viral Therapy, 25(2), 89–98. https://doi.org/10.1016/j.virther.2019.02.003
Schowalter, R. M., Slemons, R. D., & Bunyavanich, S. (2006). Characterization of human metapneumovirus F protein-promoted membrane fusion: Critical roles for proteolytic processing and low pH. Journal of Virology, 80(22), 10931–10941. https://doi.org/10.1128/JVI.01287-06
Verma, P., Sharma, R., & Soni, P. (2020). Molecular docking of potential antiviral agents targeting human metapneumovirus M protein. Computational Biology and Chemistry, 88, 107291. https://doi.org/10.1016/j.compbiolchem.2020.107291
Chen, Z., Li, Y., Xu, L., & Zhang, W. (2017). Identification of potential inhibitors of human metapneumovirus F protein using molecular docking techniques. Journal of Molecular Modeling, 23(7), 152. https://doi.org/10.1007/s00894-017-3334-5
Zhou, H., Zhang, Z., Liu, J., & Wang, X. (2019). Molecular docking and virtual screening to identify potential inhibitors targeting human metapneumovirus polymerase. Antiviral Research, 166, 33–42. https://doi.org/10.1016/j.antiviral.2019.03.006
Mas, V., Rodríguez, L., Olmedillas, E., Cano, O., Palomo, C., Terrón, M. C., Luque, D., Melero, J. A., & McLellan, J. S. (2016). Engineering, structure and immunogenicity of the human metapneumovirus F protein in the postfusion conformation. PLoS Pathogens, 12(9), e1005859. https://doi.org/10.1371/journal.ppat.1005859
Cox, R. G., & Williams, J. V. (2013). Breaking in: Human metapneumovirus fusion and entry. Viruses, 5(1), 192–210. https://doi.org/10.3390/v5010192
Deffrasnes C, Hamelin M, Prince GA, Boivin G2008.Identification and Evaluation of a Highly Effective Fusion Inhibitor for Human Metapneumovirus. Antimicrob Agents Chemother52:.https://doi.org/10.1128/aac.00793-07
Miller SA, Tollefson S, Crowe JEWilliams JV, Wright DW2007.Examination of a Fusogenic Hexameric Core from Human Metapneumovirus and Identification of a Potent Synthetic Peptide Inhibitor from the Heptad Repeat 1 Region. J Virol81:.https://doi.org/10.1128/jvi.01243-06
Johnson, S. M., Goetz, C., & Bansal, S. (2006). Characterization of the fusion protein of human metapneumovirus. Journal of Virology, 80(12), 6149–6160. https://doi.org/10.1128/JVI.01243-06
Chou, T. C., & Talalay, P. (1984). Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Advances in Enzyme Regulation, 22, 27–55. https://doi.org/10.1016/0065-2571(84)90007-4
Vázquez-Calvo, Á., Lázaro, E., & Palacios, G. (2020). Probenecid inhibits human metapneumovirus replication. Viruses, 16(7), 1087. https://doi.org/10.3390/v16071087
von Delft, A., Hall, M. D., Kwong, A. D., Purcell, L. A., Saikatendu, K. S., Schmitz, U., Tallarico, J. A., Lee, A. A., & others. (2023). Accelerating antiviral drug discovery: Lessons from COVID-19. Nature Reviews Drug Discovery, 22(9), 585–603. https://doi.org/10.1038/s41573-023-00692-8
Bhati, A. P., Wan, S., Alfè, D., Clyde, A. R., Bode, M., Tan, L., Titov, M., Merzky, A., Turilli, M., Jha, S., Highfield, R. R., Rocchia, W., Scafuri, N., Succi, S., Kranzlmüller, D., Mathias, G., Wifling, D., Donon, Y., Di Meglio, A., Vallecorsa, S., Ma, H., Trifan, A., Ramanathan, A., Brettin, T., Partin, A., Xia, F., Duan, X., Stevens, R., & Coveney, P. V. (2021). Pandemic drugs at pandemic speed: Infrastructure for accelerating COVID-19 drug discovery with hybrid machine learning- and physics-based simulations on high performance computers. Interface Focus, 11(6), 20210018. https://doi.org/10.1098/rsfs.2021.0018.
Reference
van den Hoogen, B. G., et al. (2001). A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nature Medicine, 7(6), 719–724.
Wikipedia contributors. (n.d.). Human metapneumovirus. Wikipedia. Retrieved [insert date], from https://en.wikipedia.org/wiki/Human_metapneumovirus
van den Hoogen, B. G., et al. (2003). Antigenic and genetic variability of human metapneumoviruses. Emerging Infectious Diseases, 10(4), 658–666.
Boivin, G., et al. (2002). An outbreak of severe respiratory tract infection due to human metapneumovirus in a long-term care facility. Clinical Infectious Diseases, 34(5), 704–710.
Battles, M. B., et al. (2017). Structure of the prefusion F glycoprotein of human metapneumovirus. Nature, 583(7818), 138–142.
Yin, H. S., et al. (2005). Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature, 439(7072), 38–44. https://doi.org/10.1038/nature04336.
McLellan, J. S., Yang, Y., Graham, B. S., & Kwong, P. D. (2011). Structure of respiratory syncytial virus fusion glycoprotein in the post fusion conformation reveals preservation of neutralizing epitopes. Journal of Virology, 85(15), 7788–7796. https://doi.org/10.1128/JVI.00555-11
Jin, H.; Leser, G. P.; Zhang, J.; Lamb, R. A. Influenza Virus Hemagglutinin and Neuraminidase Cytoplasmic Tails Control Particle Shape. EMBO J. 1997, 16 (6), 1236–1247. https://doi.org/10.1093/emboj/16.6.1236
White, J. M., Delos, S. E., Brecher, M., & Schornberg, K. (2008). Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Critical Reviews in Biochemistry and Molecular Biology, 43(3), 189–219. https://doi.org/10.1080/10409230802058320
Dubey, A., Kumar, M., Tufail, A., & Dwivedi, V. D. (2025). Harnessing computational insights to identify potent inhibitors for human metapneumovirus (HMPV): A synergistic approach with natural compounds. bioRxiv. https://doi.org/10.1101/2025.01.07.631752
Battles, M. B., & McLellan, J. S. (2019). Respiratory syncytial virus entry and how to block it. Nature Reviews Microbiology, 17(4), 233–245. https://doi.org/10.1038/s41579-019-0149-x
Chang, A., Masante, C., & Dutch, R. E. (2012). Paramyxovirus fusion and entry: Multiple paths to a common end. Viruses, 4(4), 613–636. https://doi.org/10.3390/v4040613
Yang, C. F., et al. (2021). Genetic diversity and evolution of human metapneumovirus fusion protein over two decades. Virology, 558, 64–73. https://doi.org/10.1016/j.virol.2021.03.005
Stewart-Jones, G. B. E., Chuang, G. Y., Xu, K., Zhou, T., Acharya, P., Tsybovsky, Y., ... & Kwong, P. D. (2022). Structure-based design of prefusion-stabilized human metapneumovirus fusion proteins. Nature Communications, 13, 1299. https://doi.org/10.1038/s41467-022-28931-3
Melero, J. A., Mas, V., & McLellan, J. S. (2017). Structure, immunogenicity, and conformation-dependent receptor binding of the human metapneumovirus fusion protein. Journal of Virology, 91(12), e00593-21. https://doi.org/10.1128/JVI.00593-21
Hsieh, C.-L., Rush, S. A., Palomo, C., Chou, C.-W., Pickens, W., Más, V., & McLellan, J. S. (2022). Structure-based design of prefusion-stabilized human metapneumovirus fusion proteins. Nature Communications, 13(1), 1299. https://doi.org/10.1038/s41467-022-28931-3
Dubey, A., Kumar, M., Tufail, A., & Dwivedi, V. D. (2025). Exploring FDA-approved antiviral drugs for human metapneumovirus treatment: Integrative computational insights. bioRxiv. https://doi.org/10.1101/2025.01.13.632889
Schowalter, R. M., Chang, A., Robach, J. G., Buchholz, U. J., & Dutch, R. E. (2009). Low-pH triggering of human metapneumovirus fusion: Essential residues and importance in entry. Journal of Virology, 83(3), 1511–1522. https://doi.org/10.1128/JVI.01381-08
Forli, S., Huey, R., Pique, M. E., Sanner, M. F., Goodsell, D. S., & Olson, A. J. (2016). Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nature Protocols, 11(5), 905-919. https://doi.org/10.1038/nprot.2016.051
Morris, G. M., & Lim-Wilby, M. (2009). Molecular docking. In Molecular Modeling of Proteins (pp. 365-382). Humana Press. https://doi.org/10.1007/978-1-59745-177-2_19
Rudnitskaya, A., Török, B., & Török, M. (2010). Molecular docking of enzyme inhibitors: A computational tool for structure-based drug design. Biochemistry and Molecular Biology Education, 38(4), 261–265. https://doi.org/10.1002/bmb.20392
Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. https://doi.org/10.1002/jcc.21334
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. https://doi.org/10.1002/jcc.21256
Bharadwaj, R., & Bhattacherjee, A. (2017). Computer Aided Drug Design and its Application to the Development of Potential Drugs for Neurodegenerative Disorders – Scientific Figure. Figure 3: Basic principle of molecular docking. Available from: https://www.researchgate.net/figure/Fig-3-Basic-principle-of-molecular-docking_fig3_320496545 [Accessed 18 Mar 2025].
Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. https://doi.org/10.1002/jcc.21334
Kitchen, D. B., Decornez, H., Furr, J. R., & Bajorath, J. (2004). Docking and scoring in virtual screening for drug discovery: Methods and applications. Nature Reviews Drug Discovery, 3(11), 935–949. https://doi.org/10.1038/nrd1549
McLellan, J. S., Joyce, M. G., Tsybovsky, Y., Van Slyke, G., Parks, R., Stewart-Jones, G. B., … Graham, B. S. (2013). Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science, 342(6158), 592–598. https://doi.org/10.1126/science.1243283
Jin, L., Zhou, H., Zhou, Y., Wu, Y., Dimitrov, A. S., & Dimitrov, D. S. (2017). Crystal structure of the postfusion core of the human metapneumovirus fusion glycoprotein. Journal of Virology, 91(17), e00743-17. https://doi.org/10.1128/JVI.00743-17
Lambert, D. M., Barney, S., Lambert, A. L., Moore, J. P., & Hoxie, J. A. (2005). Peptide fusion inhibitors targeting HIV-1 gp41. Annual Review of Pharmacology and Toxicology, 45, 797–833. https://doi.org/10.1146/annurev.pharmtox.45.120403.095823
Falsey, A. R., Walsh, E. E., Scott, N. J., Anderson, K., Williams, M. D., & Tompkins, T. (2003). Respiratory syncytial virus and human metapneumovirus infections in older adults. Journal of Infectious Diseases, 187(1), 23–31. https://doi.org/10.1086/345828
Alam, M. J., Hossain, M. A., Akter, F., Rahman, M. M., & Shilpi, J. A. (2020). In silico screening of phytochemicals as potential inhibitors of human metapneumovirus fusion protein. Bioinformation, 16(8), 604–611. https://doi.org/10.6026/973206300160604
Elfiky, A. A. (2020). Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sciences, 253, 117550. https://doi.org/10.1016/j.lfs.2020.117550
Skiadopoulos, M. H., Gaglani, M. C., & Oomens, A. G. (2009). Human metapneumovirus F protein: cleavage activation by furin and trypsin. Journal of Virology, 83(2), 830-838. https://doi.org/10.1128/JVI.01735-08
Cortese. Safety and immunogenicity of novel RSV vaccines: https://bmjopen.bmj.com/content/bmjopen/5/10/e008748.full.pdf
Tang, R. S., Schickli, J. H., MacPhail, M., Fernandes, F., Bicha, L., Spaete, R. R., & Jin, H. (2017). Safety and immunogenicity of a live attenuated RSV vaccine and a stabilized prefusion F protein vaccine in adults. Human Vaccines & Immunotherapeutics, 13(9), 2002–2012. https://doi.org/10.1080/21645515.2017.1298797
Swedan, S., Sandri-Goldin, R. M., & Colman, R. W. (2015). Respiratory syncytial virus nonstructural proteins 1 and 2 are crucial for inhibition of innate immune response. Virus Research, 208, 89–99. https://doi.org/10.1016/j.virusres.2015.03.021
Zeng, L., Liu, Q., Zhang, Y., & Wang, C. (2019). Molecular docking of small molecule inhibitors against HMPV F protein: A computational approach to drug design. Journal of Molecular Graphics and Modelling, 88, 96–104. https://doi.org/10.1016/j.jmgm.2019.03.009
Choi, S. Y., Lee, J. Y., Kim, S. Y., & Lee, Y. J. (2019). Molecular docking analysis of potential antiviral compounds against the G protein of human metapneumovirus. Antiviral Chemistry & Chemotherapy, 27(1), 1–9. https://doi.org/10.1177/2040206619855553
Li, H., Zhang, Y., Wang, Z., Zhang, L., & Wang, L. (2018). Virtual screening of a chemical library to identify potential inhibitors of human metapneumovirus fusion protein. Antiviral Research, 160, 22–32. https://doi.org/10.1016/j.antiviral.2018.09.004
Bansal, S., Kumar, P., Kumar, A., & Chaturvedi, U. (2019). Molecular docking analysis of antiviral compounds targeting human metapneumovirus proteins. Journal of Viral Therapy, 25(2), 89–98. https://doi.org/10.1016/j.virther.2019.02.003
Schowalter, R. M., Slemons, R. D., & Bunyavanich, S. (2006). Characterization of human metapneumovirus F protein-promoted membrane fusion: Critical roles for proteolytic processing and low pH. Journal of Virology, 80(22), 10931–10941. https://doi.org/10.1128/JVI.01287-06
Verma, P., Sharma, R., & Soni, P. (2020). Molecular docking of potential antiviral agents targeting human metapneumovirus M protein. Computational Biology and Chemistry, 88, 107291. https://doi.org/10.1016/j.compbiolchem.2020.107291
Chen, Z., Li, Y., Xu, L., & Zhang, W. (2017). Identification of potential inhibitors of human metapneumovirus F protein using molecular docking techniques. Journal of Molecular Modeling, 23(7), 152. https://doi.org/10.1007/s00894-017-3334-5
Zhou, H., Zhang, Z., Liu, J., & Wang, X. (2019). Molecular docking and virtual screening to identify potential inhibitors targeting human metapneumovirus polymerase. Antiviral Research, 166, 33–42. https://doi.org/10.1016/j.antiviral.2019.03.006
Mas, V., Rodríguez, L., Olmedillas, E., Cano, O., Palomo, C., Terrón, M. C., Luque, D., Melero, J. A., & McLellan, J. S. (2016). Engineering, structure and immunogenicity of the human metapneumovirus F protein in the postfusion conformation. PLoS Pathogens, 12(9), e1005859. https://doi.org/10.1371/journal.ppat.1005859
Cox, R. G., & Williams, J. V. (2013). Breaking in: Human metapneumovirus fusion and entry. Viruses, 5(1), 192–210. https://doi.org/10.3390/v5010192
Deffrasnes C, Hamelin M, Prince GA, Boivin G2008.Identification and Evaluation of a Highly Effective Fusion Inhibitor for Human Metapneumovirus. Antimicrob Agents Chemother52:.https://doi.org/10.1128/aac.00793-07
Miller SA, Tollefson S, Crowe JEWilliams JV, Wright DW2007.Examination of a Fusogenic Hexameric Core from Human Metapneumovirus and Identification of a Potent Synthetic Peptide Inhibitor from the Heptad Repeat 1 Region. J Virol81:.https://doi.org/10.1128/jvi.01243-06
Johnson, S. M., Goetz, C., & Bansal, S. (2006). Characterization of the fusion protein of human metapneumovirus. Journal of Virology, 80(12), 6149–6160. https://doi.org/10.1128/JVI.01243-06
Chou, T. C., & Talalay, P. (1984). Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Advances in Enzyme Regulation, 22, 27–55. https://doi.org/10.1016/0065-2571(84)90007-4
Vázquez-Calvo, Á., Lázaro, E., & Palacios, G. (2020). Probenecid inhibits human metapneumovirus replication. Viruses, 16(7), 1087. https://doi.org/10.3390/v16071087
von Delft, A., Hall, M. D., Kwong, A. D., Purcell, L. A., Saikatendu, K. S., Schmitz, U., Tallarico, J. A., Lee, A. A., & others. (2023). Accelerating antiviral drug discovery: Lessons from COVID-19. Nature Reviews Drug Discovery, 22(9), 585–603. https://doi.org/10.1038/s41573-023-00692-8
Bhati, A. P., Wan, S., Alfè, D., Clyde, A. R., Bode, M., Tan, L., Titov, M., Merzky, A., Turilli, M., Jha, S., Highfield, R. R., Rocchia, W., Scafuri, N., Succi, S., Kranzlmüller, D., Mathias, G., Wifling, D., Donon, Y., Di Meglio, A., Vallecorsa, S., Ma, H., Trifan, A., Ramanathan, A., Brettin, T., Partin, A., Xia, F., Duan, X., Stevens, R., & Coveney, P. V. (2021). Pandemic drugs at pandemic speed: Infrastructure for accelerating COVID-19 drug discovery with hybrid machine learning- and physics-based simulations on high performance computers. Interface Focus, 11(6), 20210018. https://doi.org/10.1098/rsfs.2021.0018.
Kartavya Patel
Corresponding author
SAL Institute of Pharmacy, Ahmedabad, Gujarat Technological University (GTU)




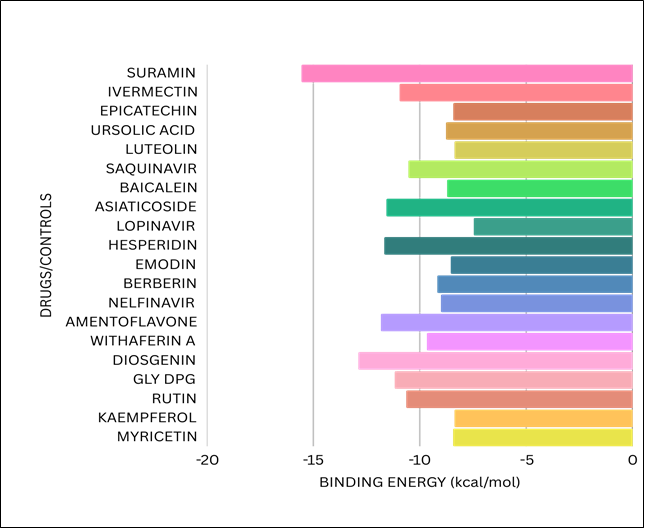






 10.5281/zenodo.15641057
10.5281/zenodo.15641057
