Dr. Moopen’s College of Pharmacy
The main aim of this research is to design and synthesize a scaffold of 2-phenyl indole derivatives and evaluate their anti-cancer activity against selected target protein EGFR Kinase protein (PDB ID: 3POZ) through various techniques, such as chemical synthesis, characterization, and in-silico modelling. The study aims to discover and evaluate novel compounds that could potentially be developed into effective anticancer drugs. The designed scaffolds were docked against EGFR kinase using Schrodinger suit software. 4 -(2-phenyl-1H indol- 1-yl) benzoic acid is synthesized by reaction between 2-Phenyl indole and para-amino benzoic acid. 2-Phenyl indole reacts with thioglycolic acid to yield [(2 -phenyl-4H-indol-4- yl) sulfanyl] acetic acid. The synthesized lead molecules were characterized by means of FTIR, Mass spectroscopic techniques. The molecule n4PI exhibited strong binding affinity with EGFR Kinase protein (PDB ID: 3POZ) with binding energy of -7 kcal/mol and thus turned out to be the most active 2-Phenyl indole derivative against EGFR Kinase protein. The synthesis of the novel 2 -phenyl indole derivatives was successful, and the compounds were characterized to confirm their chemical structures and purities.
1.1 Indole
Benzo pyrrole, which is produced by combining indigo with oleum, is known by the trivial term indole. Baeyer and Knop reduced indigo in 1866 to get two compounds, dihydroxy indole and oxindole, which they later proposed as the term "Indole." They believed that indigo was a derivative of C8H7N.Different pharmacological actions are present in heterocyclic compounds. Five or six members of an atom of nitrogen, sulphur, or oxygen are substituted and fused.[1] Heterocyclic compounds have a significant role in drug development and medical chemistry. 5-7 A fused heterocyclic compound, the indole nucleus consists of a five-membered pyrrole ring and a six-membered benzene ring. The majority of commercially accessible medications are heterocyclic molecules that contain nitrogen. In medicinal chemistry, the indole nucleus found in the derivatives plays a significant role in pharmacological actions such as anticancer, antiviral, antihypertensive, anti-inflammatory, analgesic, antibacterial, antifungal, and antidepressant properties.
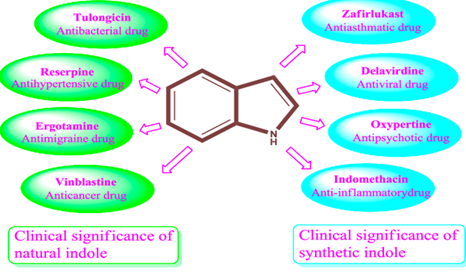
Figure 1 : Biological activity of 2-phenyl indole
Indole is a crucial neurotransmitter that contains a heterocyclic nucleus, the backbone of the natural hormone melatoin, the neuro transmitter serotonin, and the vital amino acid tryptophan. The derivatives of indole have either been synthesized or extracted from natural sources. Indole alkaloids such as vincristine, vinblastine, and mitomycin, as well as their counterparts, are utilized in antibacterial and cancer chemotherapy, respectively. They are a good starting point for the creation of novel antibacterial and antifungal drugs since they contain indole. The main aim of this research is to design and synthesize a series of 2-phenyl indole derivatives with promising anticancer activity. Through various techniques, such as chemical synthesis, characterization, and in-silico modeling, the study aims to discover and evaluate novel compounds that could potentially be developed into effective anticancer drugs.[2,3,4]
MATERIALS USED
2.1 In-silico drug design studies
2.1.1 ChemDraw
Designing a molecule using ChemDraw is a powerful tool for creating chemical structures and visualizing molecular designs. ChemDraw allows you to represent organic and inorganic molecules in various formats. It offers a user-friendly interface and a range of tools and features that facilitate the creation of high-quality chemical diagrams.[5]
2.1.2 IBM RXN chemistry for Reaction prediction
A cloud-based platform called IBM RXN for Chemistry was created by IBM and uses machine learning (ML) and artificial intelligence (AI) methods to forecast and produce chemical reactions. It is intended to help researchers and chemists find and improve synthetic pathways and reactions[6].
2.1.3 PubChem for Molecular library
The National Centre for Biotechnology Information (NCBI), a division of the National Library of Medicine in the United States, is responsible for maintaining the free online database PubChem. It offers a thorough compilation of chemical data, such as chemical structures, characteristics, biological activity, and citations to academic publications. Chemists, researchers, and the general public can explore and access chemical and biological data with the help of PubChem[7]
2.1.4 RCSB Protein Data Bank (PDB)
The RCSB Protein Data Bank (PDB) is a comprehensive and widely-used online resource that provides access to three-dimensional structures of biological macromolecules, including proteins, nucleic acids, and complex assemblies. It is a collaborative effort between the Research Collaboratory for Structural Bioinformatics (RCSB), which includes Rutgers University, the University of California, San Diego, and the University of California, San Francisco.[8]
Schrodinger Suite for Docking studies
Schrodinger Suite, developed by Schrödinger, Inc., is a comprehensive software package widely used in computational chemistry and drug discovery. It provides a range of tools and modules for molecular modeling, simulations, structure-based drug design, and data analysis.[9]
Swiss ADME
SWISS ADME (Absorption, Distribution, Metabolism, and Excretion) is a web-based tool developed by the Swiss Institute of Bioinformatics (SIB) that provides predictions and assessments of various pharmacokinetic and drug-like properties of small molecules. It aids in the early stages of drug discovery and optimization by providing insights into the potential ADME characteristics of compounds.[10,11]
2.2 Molecular Docking
2.2.1 Software’s used
Table 1 Showing list of Software used for study
|
Sr. no |
Software |
Version |
Company |
|
1. |
ChemDraw |
12.0 |
Cambridge Soft |
|
2. |
IBM Rxn chemistry |
Web application |
IBM Research |
|
3. |
Pubchem |
Webapplication |
National institutes of Health |
|
4. |
RCSB PDB |
Webapplication |
World-wide protein data bank |
|
5. |
Swiss ADME |
Webapplication |
Swiss ADME |
|
6. |
Schondinger suit |
2023-1 |
Schondinger |
2.3 Synthesis
2.3.1 Equipment’s & apparatus used
The synthesis of 2-phenyl indole can be accomplished using various methods and reagents. The specific equipment and apparatus required may vary depending on the chosen synthetic route.[12,13,14,15]
Table 2 Showing list of Laboratory Equipment used in the study
|
SI no |
Equipment’s used |
Company |
|
1. |
Melting point apparatus |
LABTRONICS |
|
2. |
Magnetic stirrer |
KEMI |
|
3. |
UV-Vis spectrophotometer |
ANTECH |
|
4. |
pH meter |
KEMI |
|
5. |
Vaccum pump |
VALUS |
|
6. |
Heating mantle |
KEMI |
|
7. |
Electronic weighing balance |
SHIMADZU |
|
8. |
Hot air oven |
KEMI |
|
9. |
Fume cupboard |
KEMI |
Table 3 Showing list of Laboratory Apparatus used for the study
|
SI no |
Apparatus used |
Quantity |
Company |
|
1. |
TLC plates, Developing chamber |
1 |
BOROSIL |
|
2. |
Thermometer |
1 |
LABWORLD |
|
3. |
Water bath |
1 |
KEMI |
|
4. |
Round-bottom flask |
1 |
BOROSIL |
|
5. |
Condenser |
1 |
BOROSIL |
|
6. |
Separatory funnel |
1 |
BOROSIL |
|
7. |
Beaker |
3 |
BOROSIL |
|
8. |
Funnel |
2 |
BOROSIL |
|
9. |
Glass rod |
3 |
BOROSIL |
|
10. |
Watch glass |
2 |
BOROSIL |
|
11. |
Measuring cylinder |
2 |
BOROSIL |
|
12. |
Test tubes |
1 |
BOROSIL |
|
13. |
Conical flask |
1 |
BOROSIL |
|
14. |
Pipettes |
4 |
BOROSIL |
|
15. |
Distillation apparatus |
1 |
BOROSIL |
METHODS
Scheme Preparation
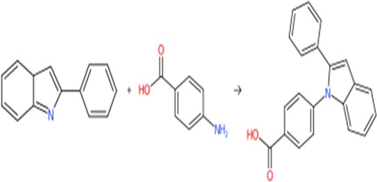
Figure 2 : Scheme I
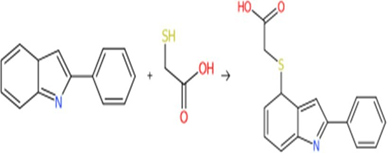
Figure 3: Scheme II
Scheme I: Reaction with 2-Phenyl indole: 2-Phenyl indole reacts with Para amino benzoic acid to yield 4 -(2-phenyl-1H indol- 1-yl) benzoic acid(4PI)
Scheme II: 2-Phenyl indole reacts with Thioglycolic acid to yield [(2 -phenyl-4H-indol-4- yl)sulfanyl]acetic acid.
Scheme I
Step-1: Preparation of Acetophenone phenyl hydrazone[16,17,18] 5.15 g of acetophenone (0.042mol) was taken in a conical flask containing 5 mL of Ethanol and 1 mL of glacial acetic acid. To the above mixture 4.53 g of phenyl hydrazine was added dropwise with constant swirling. Heat the reaction mixture in sand bath for 10 minutes and cool the resulting mixture in ice bath, allow the product to precipitate. Collect the precipitate by filtration on filter paper and wash with dilute hydrochloric acid (3 mL) followed by cold ethanol (5 mL). Allow the precipitate to air dry on the filter paper and recrystallized to get pure product from ethanol.
Step-2: Preparation of 2-Phenyl indole
Place crude acetophenone phenyl hydrazone beaker containing 15 mL of ortho phosphoric acid and 5 ml. of concentrated sulphuric acid. Heat the resulting mixture on the water bath for 20 minutes at 100-120°C with constant stirring. Pour the hot reaction mixture into 50 mL of cold water and wash the beaker with few mL of water. Collect the precipitated crude product on a filter paper, wash it with ethanol and allow it to air dry on the filter paper. Recrystallize the crude product from ethanol-water, using about g of decolorizing carbon and filter hot calculate the yield and obtain melting point.
Step-3: preparation of 4-(2-phenyl-1H-indol-1-yl) benzoic acid
A mixture of 2-phenyl indole (1.89g 0.01mol) and Para amino benzoic acid (1.37g 0.01mol) in ethanol (40ml) were refluxed for 4hours and excess of solvent was removed by distillation process. The resulting solid was dried and recrystalised from dilute ethanol to obtain the product.
SPECTRAL ANALYSIS: The synthesized derivatives were characterized using advanced analytical techniques like, Nuclear Magnetic Resonance (NMR) spectroscopy & Mass Spectrometry (MS)
RESULTS & DISCUSSION
5.1 In Silico Drug Design: The initial phase of the study involved the utilization of in silico techniques to design and predict the properties of novel 2-phenyl indole derivatives with potential therapeutic effects. Computational tools such as molecular docking, virtual screening, and pharmacokinetic analysis were employed to identify potential drug candidates. The results of the Insilco drug design phase revealed several compounds with favourable binding affinities and drug-like properties, suggesting their potential as effective drug molecules.
|
Sr. No. |
Code |
Compound |
Docking score |
No of H- bonding |
|
1. |
1PI |
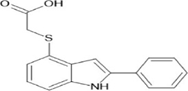
[(2-phenyl-2,3-dihydro-1H-indol-4-yl)sulfanyl]acetic acid |
-6 |
4 |
|
2. |
3PI |
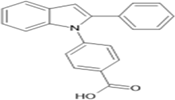
4-(2-phenyl-1H-indol-1-yl)benzoic acid |
-7 |
3 |
|
3. |
n5PI |
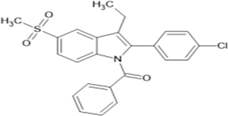
[2-(4-chlorophenyl)-3-ethyl-5-(methanesulfonyl)-1H indol-1-yl](phenyl)methanone |
-6 |
7 |
|
4. |
n4PI |
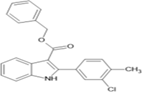
benzyl 2-(3-chloro-4-methylphenyl)-2,3-dihydro-1H indole-3-carboxylate |
-7 |
1 |
|
5. |
n7PI |
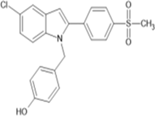
4-({5-chloro-2-[4-(methanesulfonyl)phenyl]-1H-indol-1- yl}methyl)phenol |
-5 |
5 |
|
6. |
n8PI |
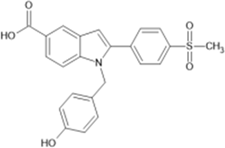
1-[(4-hydroxyphenyl)methyl]-2-[4- (methanesulfonyl)phenyl]-1H-indole-5-carboxylic acid |
-5 |
8 |
|
7. |
25PI |
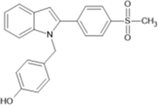
4-({2-[4-(methanesulfonyl)phenyl]-1H-indol-1- yl}methyl)phenol |
-7 |
5 |
Standard drug
|
8. |
STD |
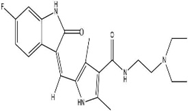
N-[2-(diethylamino)ethyl]-5-[(Z)-(5-fluoro-2-oxo-1H- indol-3-ylidene)methyl]-2,4-dimethyl-1H-pyrrole-3- carboxamide |
-5 |
|
Result showing Physichochemical Properties, Lipophilicity, Druglikeness, Water Solubility, Pharmacokinetics, Medicinal Chemistry Parameter for each newly developed derivatives.
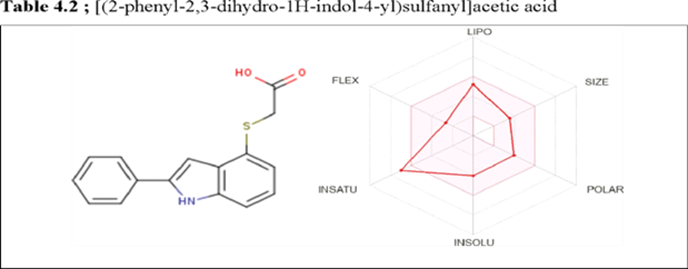
|
SMILE : OC(=O)CSc1cccc2c1CC(N2)c1ccccc1 |
|||
|
Physicochemical Properties |
Water Solubility |
||
|
Formula |
C16H15NO2S |
Log S (ESOL) |
-4.04 |
|
Molecular weight |
285.36 g/mol |
Solubility |
2.61e-02 mg/ml ; 9.16e-05 mol/l |
|
Num. heavy atoms |
20 |
Class |
Moderately Soluble |
|
Arom, heavy atoms |
12 |
Log S (Ali) |
-4.82 |
|
Fraction Csp3 |
0.19 |
Solubility |
4.29e-03 mg/ml ; 1.50e-05 mol/l |
|
Rotatable bonds |
4 |
Class |
Moderately Soluble |
|
H-bond acceptors |
2 |
LogS(SILICOS- IT) |
-5.17 |
|
H-bond donors |
2 |
Solubility |
1.92e-03 mg/ml ; 6.75e-06 mol/l |
|
Molar Refractivity |
84.32 |
Class |
Moderately Soluble |
|
Lipophilicity |
Druglikeness |
||
|
Log Po/w (iLOGP) |
2.06 |
Lipinski |
Yes; 0 Violation |
|
Log Po/w (XLOGP3) |
3.57 |
Ghose |
Yes |
|
Log Po/w (WLOGP) |
2.68 |
Veber |
Yes |
|
Log Po/w (MLOGP) |
2.91 |
Egan |
Yes |
|
Log Po/w (SILICOS) |
3.15 |
Muegge |
Yes |
|
Consensus Log Po/w |
2.87 |
Bioavailability |
0.85 |
|
Pharmacokinetics |
Medicinal Chemistry |
||
|
GI absorption |
High |
PAINS |
0 alert |
|
BBB permeant |
Yes |
Brenk |
0 alert |
|
P-gp substrate |
Yes |
Leadlikeness |
No; 1 violation: XLOGP3>3.5 |
|
CYP1A2 inhibitor |
Yes |
Synthetic accessibility |
2.99 |
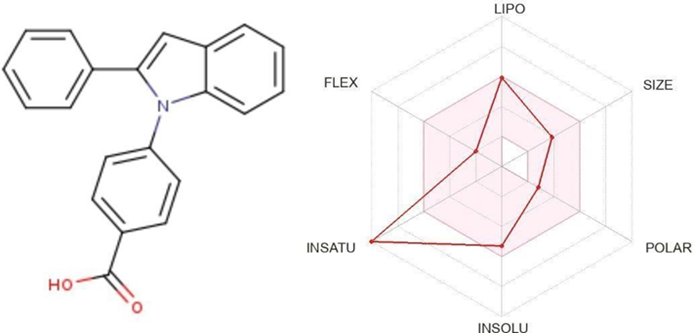
Table 4.3 ; 4-(2-phenyl-1H-indol-1-yl)benzoic acid
|
|
|||
|
SMILE : OC(=O)c1ccc(cc1)n1c(cc2c1cccc2)c1ccccc1 |
|||
|
Physicochemical Properties |
Water Solubility |
||
|
Formula |
C21H15NO2 |
Log S (ESOL) |
-5.29 |
|
Molecular weight |
313.35 g/mol |
Solubility |
1.62e-03 mg/ml ; 5.16e-06 mol/ |
|
Num. heavy atoms |
24 |
Class |
Moderately soluble |
|
Arom, heavy atoms |
21 |
Log S (Ali) |
-5.47 |
|
Fraction Csp3 |
0.00 |
Solubility |
1.06e-03 mg/ml ; 3.38e-06 mol/l |
|
Rotatable bonds |
3 |
Class |
Moderately soluble |
|
H-bond acceptors |
2 |
LogS(SILICOS- IT) |
-6.85 |
|
H-bond donors |
1 |
Solubility |
4.45e-05 mg/ml ; 1.42e-07 mol/l |
|
Molar Refractivity |
95.67 |
Class |
Poorly soluble |
|
Lipophilicity |
Druglikeness |
||
|
Log Po/w (iLOGP) |
2.61 |
Lipinski |
Yes; 0 violation |
|
Log Po/w (XLOGP3) |
4.85 |
Ghose |
Yes |
|
Log Po/w (WLOGP) |
5.00 |
Veber |
Yes |
|
Log Po/w (MLOGP) |
3.98 |
Egan |
Yes |
|
Log Po/w (SILICOS) |
4.00 |
Muegge |
Yes |
|
Consensus Log Po/w |
4.09 |
Bioavailability |
0.85 |
|
Pharmacokinetics |
Medicinal Chemistry |
||
|
GI absorption |
High |
PAINS |
0 alert |
|
BBB permeant |
Yes |
Brenk |
0 alert |
|
P-gp substrate |
No |
Leadlikeness |
No; 1 violation: XLOGP3>3.5 |
|
CYP1A2 inhibitor |
Yes |
Synthetic accessibility |
2.30 |
Table 4.4 ;benzyl 2-(3-chloro-4-methylphenyl)-2,3-dihydro-1H-indole-3- carboxylate
|
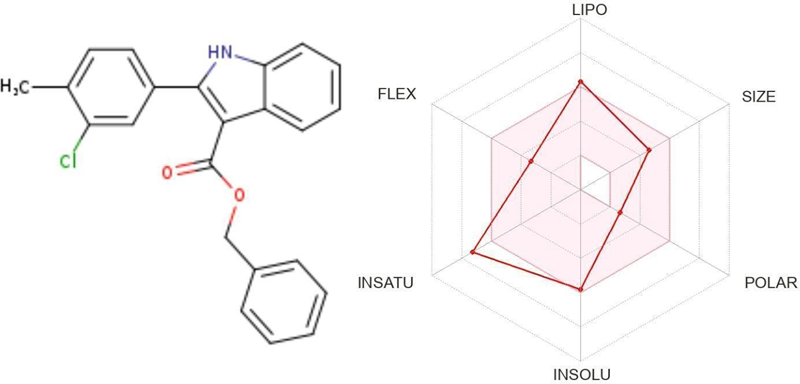
|
|||
|
SMILE : O=C(C1C(Nc2c1cccc2)c1ccc(c(c1)Cl)C)OCc1ccccc1 |
|||
|
Physicochemical Properties |
Water Solubility |
||
|
Formula |
C23H20ClNO2 |
Log S (ESOL) |
-5.82 |
|
Molecular weight |
377.86 g/mol |
Solubility |
5.67e-04 mg/ml ; 1.50e- 06 mol/l |
|
Num. heavy atoms |
27 |
Class |
Moderately soluble |
|
Arom, heavy atoms |
18 |
Log S (Ali) |
-6.08 |
|
Fraction Csp3 |
0.17 |
Solubility |
3.11e-04 mg/ml ; 8.24e- 07 mol/l |
|
Rotatable bonds |
5 |
Class |
Poorly soluble |
|
H-bond acceptors |
2 |
LogS(SILICOS- |
-8.61 |
|
|
|
IT) |
|
|
H-bond donors |
1 |
Solubility |
9.28e-07 mg/ml ; 2.45e- 09 mol/l |
|
Molar Refractivity |
111.38 |
Class |
Poorly soluble |
|
Lipophilicity |
Druglikeness |
||
|
Log Po/w (iLOGP) |
3.68 |
Lipinski |
Yes; 1 violation: MLOGP>4.15 |
|
Log Po/w (XLOGP3) |
5.52 |
Ghose |
Yes |
|
Log Po/w (WLOGP) |
4.59 |
Veber |
Yes |
|
Log Po/w (MLOGP) |
4.50 |
Egan |
Yes |
|
Log Po/w (SILICOS) |
5.40 |
Muegge |
No; 1 violation: XLOGP3>5 |
|
Consensus Log Po/w |
4.74 |
Bioavailability |
0.55 |
|
Pharmacokinetics |
Medicinal Chemistry |
||
|
GI absorption |
High |
PAINS |
0 alert |
|
BBB permeant |
Yes |
Brenk |
0 alert |
|
P-gp substrate |
Yes |
Leadlikeness |
No; 2 violations: MW>350, XLOGP3>3.5 |
|
CYP1A2 inhibitor |
Yes |
Synthetic accessibility |
3.55 |
Table4.5; [2-(4-chlorophenyl)-3-ethyl-5-(methanesulfonyl)-1H-indol-1- yl](phenyl)methanone
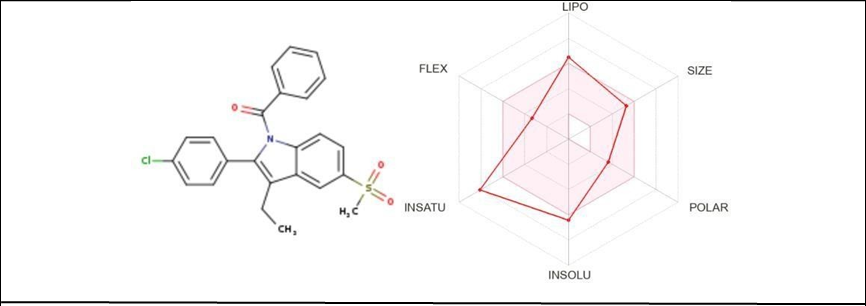
|
SMILE : CCc1c(c2ccc(cc2)Cl)n(c2c1cc(cc2)S(=O)(=O)C)C(=O)c1ccccc1 |
|||
|
Physicochemical Properties |
Water Solubility |
||
|
Formula |
C24H20ClNO3S |
Log S (ESOL) |
-6.44 |
|
Molecular weight |
437.94 g/mol |
Solubility |
1.59e-04 mg/ml ; 3.62e- 07 mol/l |
|
Num. heavy atoms |
30 |
Class |
Poorly soluble |
|
Arom, heavy atoms |
21 |
Log S (Ali) |
-7.00 |
|
Fraction Csp3 |
0.12 |
Solubility |
4.41e-05 mg/ml ; 1.01e- 07 mol/l |
|
Rotatable bonds |
5 |
Class |
Poorly soluble |
|
H-bond acceptors |
3 |
LogS(SILICOS- IT) |
-9.05 |
|
H-bond donors |
0 |
Solubility |
3.92e-07 mg/ml ; 8.95e- 10 mol/l |
|
Molar Refractivity |
121.42 |
Class |
Poorly soluble |
|
Lipophilicity |
Druglikeness |
||
|
Log Po/w (iLOGP) |
3.31 |
Lipinski |
Yes; 1 violation: MLOGP>4.15 |
|
Log Po/w (XLOGP3) |
5.87 |
Ghose |
No; 1 violation: WLOGP>5.6 |
|
Log Po/w (WLOGP) |
6.70 |
Veber |
Yes |
|
Log Po/w (MLOGP) |
4.92 |
Egan |
No; 1 violation: WLOGP>5.88 |
|
Log Po/w (SILICOS) |
5.36 |
Muegge |
No; 1 violation: XLOGP3>5 |
|
Consensus Log Po/w |
5.23 |
Bioavailability |
0.55 |
|
Pharmacokinetics |
Medicinal Chemistry |
||
|
GI absorption |
High |
PAINS |
0 alert |
|
BBB permeant |
No |
Brenk |
0 alert |
|
P-gp substrate |
No |
Leadlikeness |
No; 2 violations: MW>350, |
|
|
|
|
XLOGP3>3.5 |
|
CYP1A2 inhibitor |
No |
Synthetic accessibility |
3.07 |
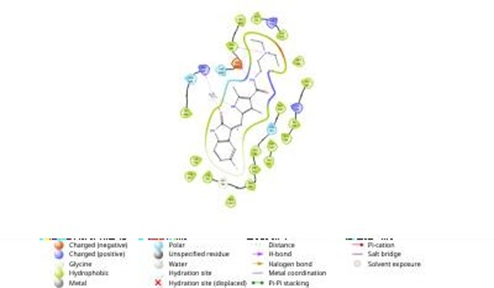
4.2 Reference compound sunitinib in the binding site of EGFR Kinase
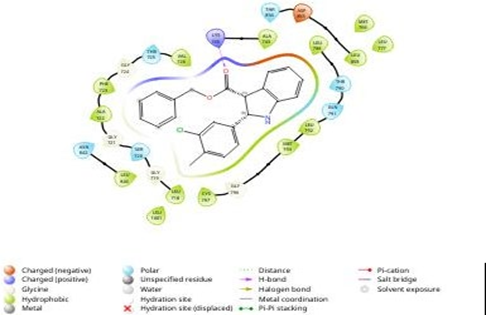
Fig 4.3 showing binding of n4PI with Amino acids of EGFR Kinase
By analyzing the docking studies 3PI and n4PI molecule was found to be the better molecule among all other molecules. The n4PI molecule exhibited strong binding affinity with EGFR Kinase protein (PDB ID: 3POZ) with binding energy of -7 kcal/mol and thus turned out to be the most active 2-Phenyl indole derivative against EGFR Kinase protein. The results of the Insilco drug design phase demonstrated the successful utilization of computational techniques in identifying potential drug candidates. The selected compounds exhibited favourable binding affinities towards the target receptors, suggesting their potential therapeutic effects. The Insilco predictions served as a guide for the subsequent synthesis and evaluation of the compounds. The synthesis of the novel 2-phenyl indole derivatives was achieved through a well-established synthetic pathway.[19,20]
SYNTHESIS
Synthesis of Novel 2-Phenyl Indole Derivatives: Following the in-silico drug design, the selected compounds were synthesized using standard organic synthesis techniques. The synthetic pathway involved the modification of the indole core structure by introducing various substituents at different positions on the phenyl ring. The synthetic reactions were carried out under controlled conditions, and the desired compounds were obtained in good yields. The compound with high docking score (-7kcal/mol) 4-(2- phenyl-1H-indol-1-yl) benzoic acid were synthesised in Laboratory.[21,22] The melting point of the compounds were determined with the aid of Digital melting point apparatus.
Table 4.6. Melting point of Synthesized compound
|
Sr no. |
Compound |
Melting Point |
|
1. |
2-Phenyl indole |
190 °C |
|
2. |
4-(2-phenyl-1H-indol-1-yl) benzoic acid |
154 °C |
Spectral analysis
Mass Spectra
The molecular ion peak appeared at m/z value is 313g/mol, corresponding to the molecular weight of the compound. The molecule 4-(2-phenyl-1 H–indole–1-yl ) benzoic acid has indole ring, phenyl group and benzoic acid group.[23]
Fragmentation pattern
313 - 77 (phenyl group) = 237 Da
Because of the presence of nitrogen atom in the indole ring the compound is highly stable and forms base peak at 237 Da.
313 - 121 (Benzoic acid) = 194 Da
FTIR Spectra: Based on the FTIR data and the known structure of the compound, we can make some observations: The presence of peaks around 1600 cm^-1 suggests the presence of C-O stretching vibrations, which align with the carbonyl group in the aromatic ring of the indole moiety. The presence of peaks around 1309.03 cm^-1 and 1128.37 cm^-1 could indicate the C-N stretching vibrations in the indole ring. Peaks around 3000 cm^-1 and 3100 cm^-1 might correspond to various C-H bending and stretching vibrations in the aromatic rings and aliphatic groups. The peaks around 3484.72 cm^-1 suggest the presence of O-H stretching vibrations, which could be due to the hydroxyl group in the carboxylic acid or possibly hydrogen bonding interactions. Peaks in the range of 2544.14 cm^-1 and 1907.60 cm^- 1 might correspond to various functional groups like C≡C stretching vibrations and C≡N stretching vibrations. Overall, the observed peaks in the FTIR data do align with the expected functional groups present in 4-(2-phenyl-1H-indol-1-yl) benzoic acid.

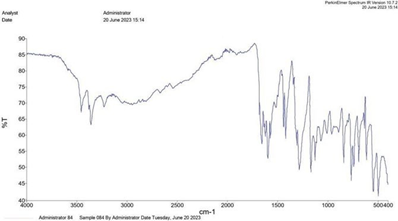
FTIR Spectrum of 4-(2-phenyl-1H-indol-1-yl) benzoic acid
|
Sr no. |
Functional group |
Reference value |
Observed value |
|
1. |
C=O |
1642cm^-1 (Instrumental method of analysis, Pavia (3rd edition)) |
1600 cm^-1 |
|
2. |
-OH |
3300cm^-1 (Instrumental method of analysis, Pavia (3rd edition)) |
3484.72 cm^-1 |
|
3. |
C-H |
3074 cm^-1(Instrumental method of analysis, Pavia (3rd edition)) |
3000 cm^-1 and 3100 cm^-1 |
|
4. |
C-N |
1224.31cm^-1(Instrumental method of analysis, Pavia (3rd edition)) |
1309.03 cm^-1 and 1128.37 cm^-1 |
CONCLUSION:
The results obtained from this study pave the way for further investigations into the therapeutic applications of the synthesized compounds. The potential demonstrated in the molecular docking studies suggests that these compounds may hold promise for the development of novel therapeutics, particularly in the areas of cancer treatment. Future studies should focus on expanding the scope of in-vitro evaluations, as well as conducting in-vivo studies to assess the compounds' efficacy and safety profiles[24]. Overall, the integration of synthesis, characterisation and insilico drug design provides a robust approach for the discovery and evaluation of novel drug candidates. The successful outcome of this study opens up possibilities for the development of new and effective treatments for various diseases, thereby contributing to the advancement of pharmaceutical research and drug discovery.[25]
REFERENCES


Swathy Lakshmi N, Dr. Lal Prasanth M L, Anzu Varghese, Safana Manakkal, Muhammed Akthar, In Silico Drug Design, Molecular Docking, Synthesis and Characterization of 2-Phenyl Indole Derivatives, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 35-48. https://doi.org/10.5281/zenodo.15780096
 10.5281/zenodo.15780096
10.5281/zenodo.15780096
