New Montfort Institute of Pharmacy Ashti Dist. Wardha.
Ivermectin is a widely used antiparasitic drug, administered globally for the management of helminthic and ectoparasitic infections (WHO, 2015). However, it’s extremely poor solubility classifies it as a BCS Class II drug, resulting in dissolution-limited absorption (Sweetman, 2017). The aim of the present study was to improve its aqueous solubility using solid dispersion techniques with PEG-6000 as the carrier polymer (Serajuddin, 1999). Solid dispersions were prepared using the fusion method in varying ratios (1:1–1:5), and the 1:3 ratio demonstrated maximum solubility enhancement (266.10 µg/mL), compared to the pure drug (3.77 µg/mL). Optimized solid dispersion was evaluated via FTIR, DSC, XRD, and SEM, confirming reduced crystallinity and absence of drug–polymer incompatibility (Sekiguchi & Obi, 1961). Dispersible tablets formulated using direct compression (F1–F4) were evaluated for standard parameters including hardness, friability, wetting time, water absorption, disintegration, and dissolution. Formulation F3 exhibited optimal qualities: hardness of 4.3 kg/cm², friability of 0.554%, disintegration time of 18 seconds, and drug release of 89.74% at 90 minutes (Ravi, 2020). Accelerated stability studies for 90 days confirmed no significant physicochemical changes, indicating formulation robustness under ICH conditions (ICH, 2003). The study concludes that PEG-6000–based solid dispersions significantly enhance the solubility and dissolution of Ivermectin, while dispersible tablets provide an improved patient-compliant dosage form for pediatric, geriatric, and dysphagic patients.
Ivermectin is a macrocyclic lactone derived from avermectins, discovered through fermentation of Streptomyces avermitilis (Campbell, 2016). It has been recognized as one of the most impactful antiparasitic drugs, with broad activity against nematodes and arthropods (WHO, 2015). Despite its clinical utility, its pharmacokinetic limitations particularly poor aqueous solubility continue to challenge oral formulation development.
Due to its hydrophobic structural nature, Ivermectin exhibits water solubility as low as 3–4 µg/mL, leading to insufficient dissolution in gastrointestinal fluids. This places it within the Biopharmaceutical Classification System (BCS) Class II, characterized by high permeability but low solubility, where dissolution is the rate-limiting step of absorption (Amidon et al., 1995). Consequently, formulation strategies for such drugs must prioritize solubility enhancement.
1.1 Necessity for Solubility Enhancement
More than 40% of newly discovered drugs suffer from poor solubility, which directly limits their bioavailability (Lipinski, 2000). For Ivermectin, which is intended for systemic therapeutic effect, improving dissolution is critical to achieving consistent blood levels (Sweetman, 2017). Solubility enhancement directly enhances oral absorption according to the Noyes–Whitney equation (Aulton & Taylor, 2018).
1.2 Solid Dispersion Technology
Solid disperion is one of the most effective methods for enhancing solubility, involving molecular dispersion of a hydrophobic drug into a hydrophilic polymer matrix (Serajuddin, 1999). The first scientific rationale for solid dispersion was introduced by Sekiguchi and Obi (1961), who demonstrated that eutectic mixtures could significantly increase drug dissolution.
Solid dispersions enhance solubility through:
1.3 PEG-6000 as a Hydrophilic Carrier
Polyethylene glycols (PEGs) are widely used in solid dispersions due to their excellent solubilizing and wetting properties (Khan et al., 2016).
PEG-6000 is specifically chosen because:
The fusion method used in the present work allows homogeneous distribution of drug molecules within the melted polymer.
1.4 Dispersible Tablets
Dispersible tablets rapidly disintegrate in a small volume of water or saliva, forming a uniform dispersion suitable for swallowing (Aulton & Taylor, 2018).
They are preferred for:
1.5 Aim of the Present Research
The objectives of this study were to:
1. Formulate solid dispersions of Ivermectin using PEG-6000.
2. Identify the ratio that provides optimal solubility and dissolution.
3. Characterize the optimized dispersion using FTIR, DSC, XRD, and SEM.
4. Formulate dispersible tablets via direct compression.
5. Evaluate their physicochemical and mechanical properties.
6. Conduct stability studies under ICH accelerated conditions.
This integrated approach enables development of an efficient, patient-friendly dosage form of Ivermectin with enhanced dissolution behavior.
2. LITERATURE REVIEW
2.1 Introduction to Ivermectin and Its Global Impact
Ivermectin, a semi-synthetic derivative of avermectins, was first introduced as a veterinary antiparasitic agent before being approved for human use in treating onchocerciasis and other parasitic diseases (Campbell, 2016). Its discovery marked a major milestone in parasitology and global public health, earning the Nobel Prize for Medicine in 2015 (WHO, 2015). The drug has been used extensively in mass drug administration (MDA) programs in Africa, Asia, and Latin America, showcasing significant safety and efficacy (Richards et al., 2018).
Despite its success, Ivermectin’s poor aqueous solubility remains a critical barrier to achieving optimal therapeutic efficacy when administered orally. The molecule is highly lipophilic, with a molecular weight exceeding 870 Da, making it inherently insoluble in water (Sweetman, 2017). Its aqueous solubility of approximately 3–4 μg/mL contributes to its poor dissolution characteristics, ultimately reducing oral absorption and leading to variable pharmacokinetics.
2.2 Solubility Challenges in BCS Class II Drugs
The Biopharmaceutics Classification System (BCS) categorizes drugs based on solubility and intestinal permeability (Amidon et al., 1995). Ivermectin falls under BCS Class II, meaning it possesses:
For such drugs, enhancing solubility is the primary determinant of improving oral bioavailability. Numerous studies emphasize that more than 40% of drugs in development suffer from poor solubility (Lipinski, 2000). Therefore, solubility enhancement remains a major focus of formulation research.
2.3 Solid Dispersion Technology in Solubility Enhancement
Solid dispersion technology has emerged as an effective strategy for enhancing the dissolution profile of poorly soluble drugs (Chiou & Riegelman, 1971). Solid dispersions typically consist of a hydrophobic drug molecularly dispersed within a hydrophilic polymer matrix, improving:
The concept was first introduced by Sekiguchi and Obi (1961), who prepared eutectic mixtures of sulfathiazole with urea. Their pioneering work established the foundation for current solid dispersion research and applications.
2.3.1 Mechanisms of Solubility Enhancement
Solid dispersions improve solubility through several mechanisms:
1. Reduction in crystallinity: Conversion of the crystalline drug into an amorphous form enhances solubility (Jain et al., 2012).
2. Improved wetting: Hydrophilic carriers such as PEG reduce interfacial tension, increasing drug–solvent contact (Serajuddin, 1999).
3. Molecular dispersion: Drugs dispersed at the molecular level dissolve faster (Vasconcelos et al., 2007)
4. Increased porosity: Greater surface irregularities improve dissolution (Khan et al., 2016).
2.4 Use of PEG-6000 in Solid Dispersions
Polyethylene glycol (PEG) polymers, especially PEG-6000, are widely used carriers for solid dispersion systems due to their favorable pharmaceutical characteristics.
2.4.1 Advantages of PEG-6000
PEG-6000 forms hydrogen bonds with drug molecules, improving their amorphization and solubility (Serajuddin, 1999).
2.5 Dispersible Tablets: A Modern Patient-Friendly Dosage Form
Dispersible tablets disintegrate rapidly in water or saliva, forming a uniform dispersion suitable for swallowing (Aulton & Taylor, 2018). These dosage forms are especially helpful for:
WHO (2015) also recommends dispersible formulations for treating parasitic infections in children who cannot swallow conventional tablets.
2.5.1 Mechanism of Disintegration in Dispersible Tablets
Disintegration occurs due to:
2.6 Previous Research Supporting Solid Dispersions and Fast-Dispersing Tablets
Several studies validate the effectiveness of solid dispersion and dispersible tablets:
Hence, the combination of solid dispersion + dispersible tablet design is scientifically justified and supported by multiple studies.
3. MATERIALS AND METHODS
3.1 MATERIALS
All chemicals were pharmaceutically graded and used without further purification.
3.2 METHODS
3.2.1 Preparation of Solid Dispersions (Fusion Method)
Solid dispersions were prepared in drug-to-carrier ratios ranging from 1:1 to 1:5.
Procedure:
1. PEG-6000 was weighed and melted at 60–70°C.
2. Ivermectin was added slowly with continuous stirring.
3. The molten mixture was cooled to room temperature.
4. Solid mass was ground and sieved (mesh #60).
5. Stored in desiccators for further use.
This fusion method is widely supported in literature (Serajuddin, 1999; Singh et al., 2016).
3.2.2 Solubility Studies (with Citation)
Solubility of pure drug and solid dispersion was assessed in:
Distilled water
0.1 N HCl
Methanol
Method was adapted from Higuchi & Connors (1965).
Solid dispersion 1:3 showed maximum solubility: 266.10 μg/mL.
3.2.3 Fourier Transform Infrared Spectroscopy (FTIR)
FTIR spectra were recorded to determine drug–polymer interactions.
Absence of new peaks indicates compatibility (Jain et al., 2012).
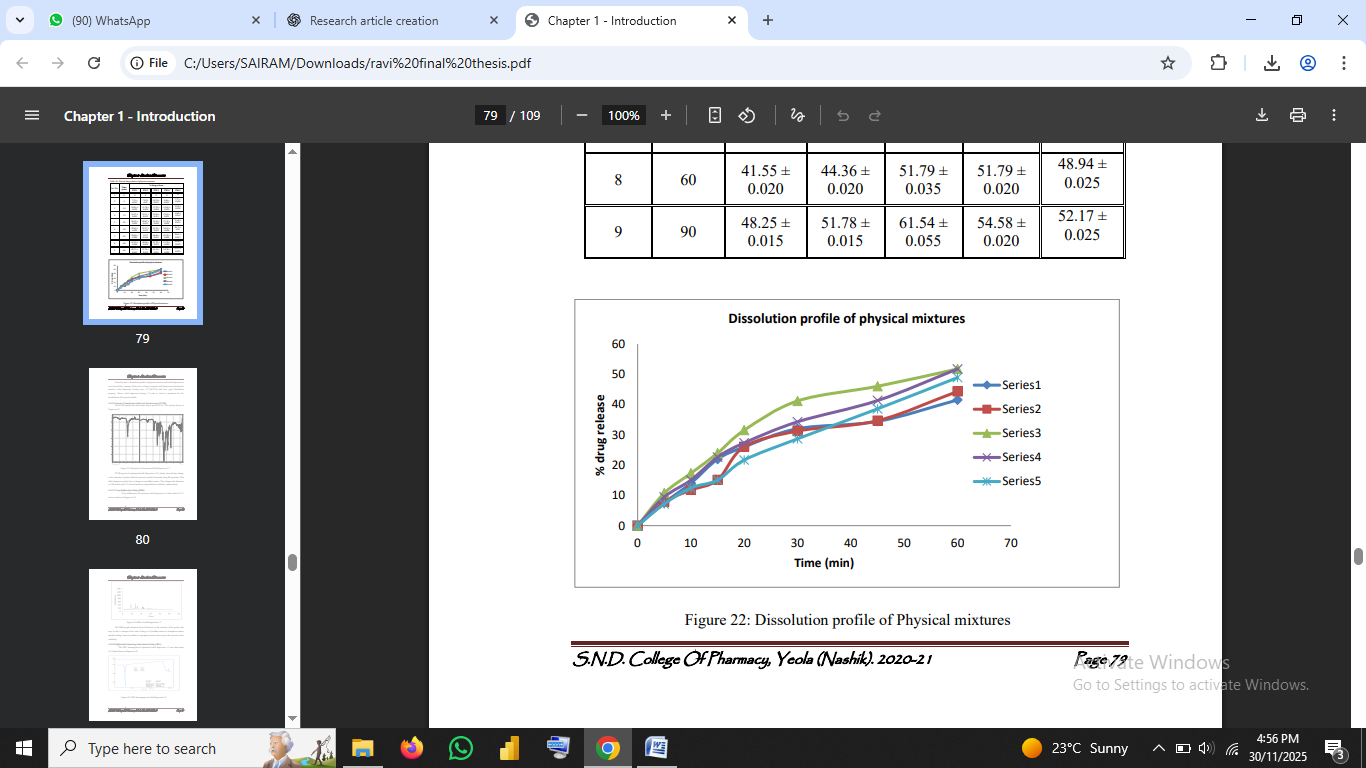
Fig no.1: FTIR spectrum of pure drug and optimized solid dispersion
3.2.4 Differential Scanning Calorimetry (DSC)
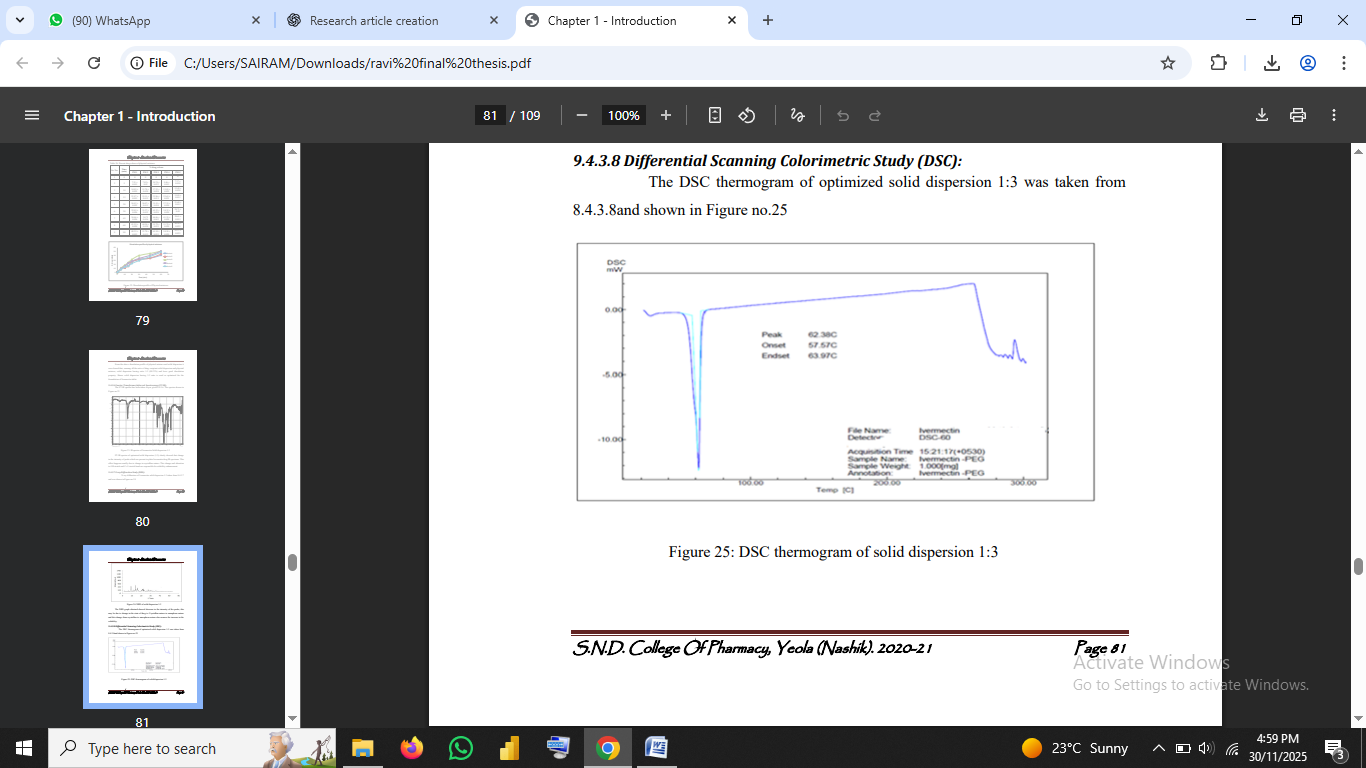
Fig no.2: DSC thermogram of solid dispersion 1:3
3.2.5 X-ray Diffraction (XRD)
XRD patterns were recorded to observe crystallinity.
Reduced peak intensity in solid dispersion indicated amorphous conversion (Vasconcelos et al., 2007).
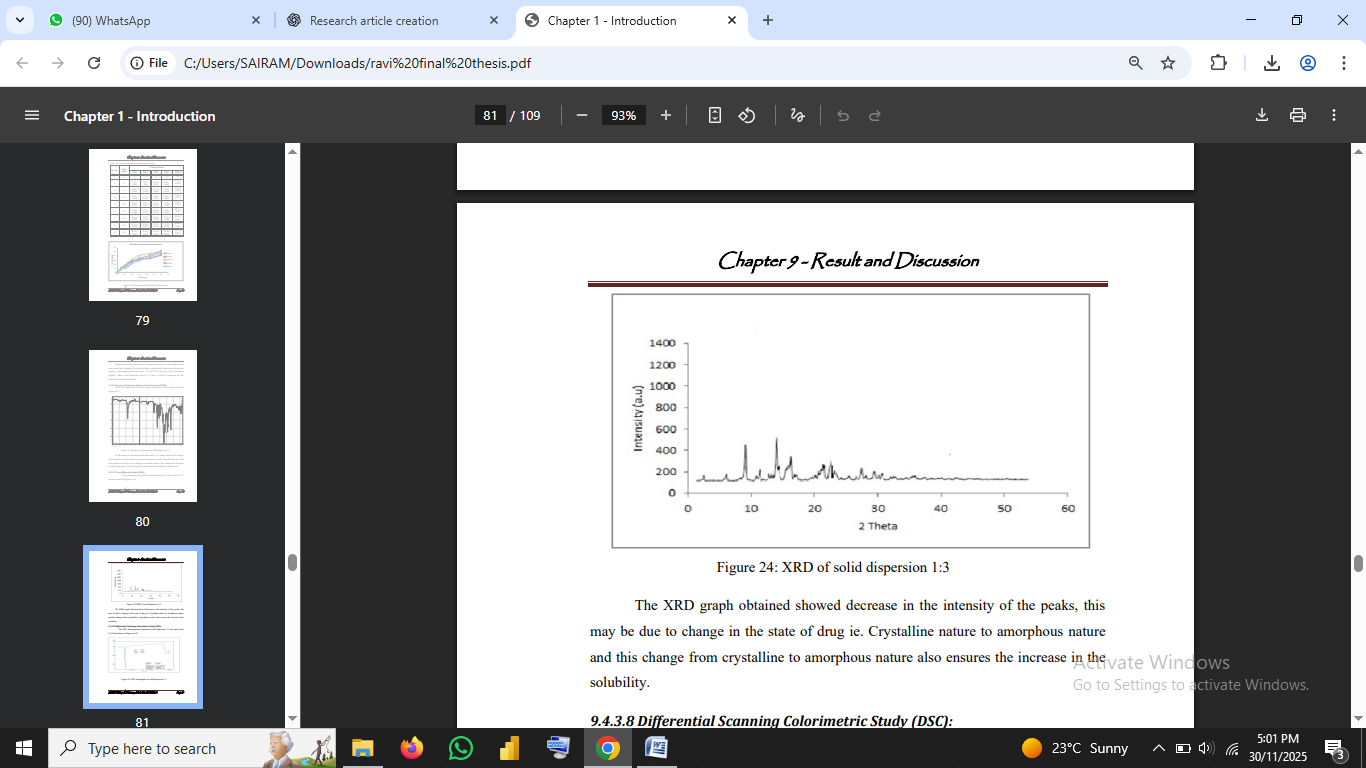
Fig no3: XRD of solid dispersion 1:3
3.2.6 Scanning Electron Microscopy (SEM):
SEM images illustrate morphological changes.
Pure drug showed crystalline particles; solid dispersion particles appeared smoother and amorphous.
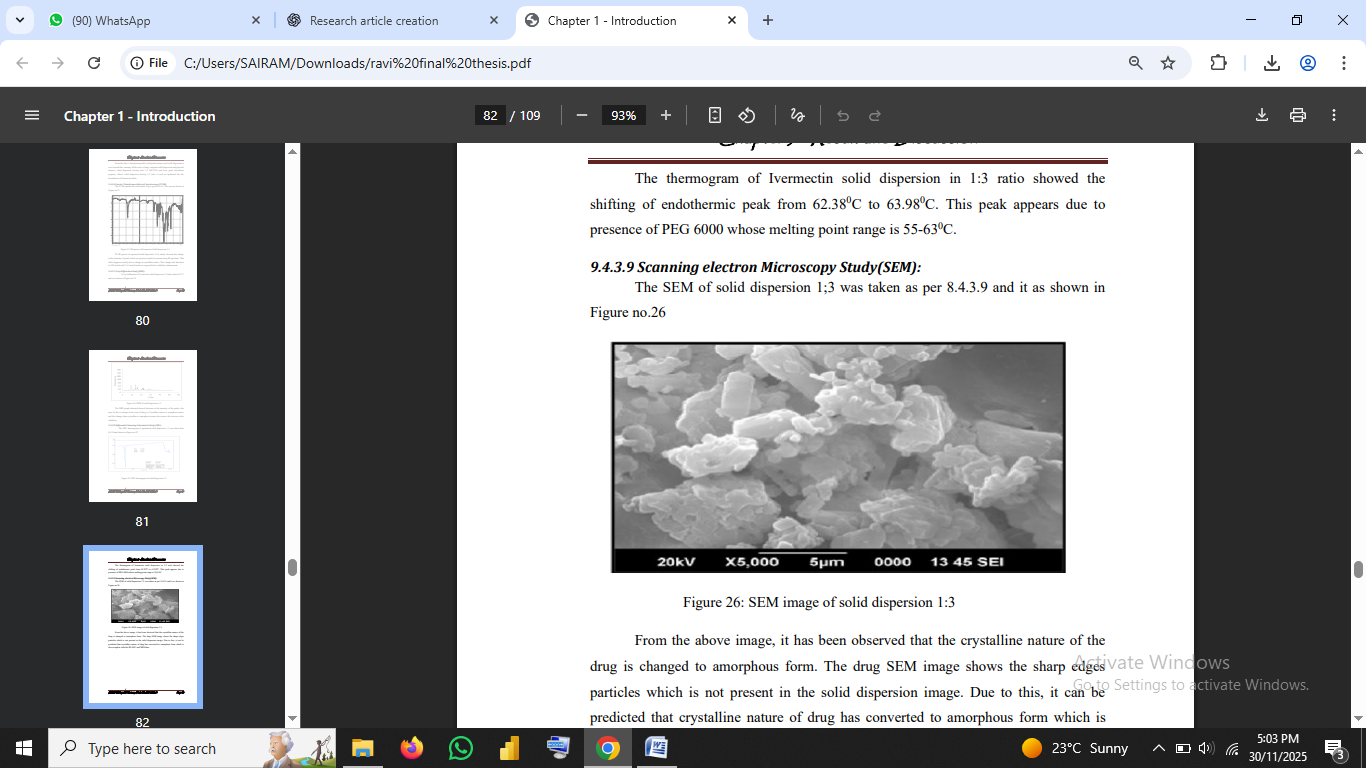
Fig no.4: SEM image of solid dispersion 1:3
3.2.7 Tablet Formulation (F1–F4)
Tablets were prepared via direct compression, a widely accepted technique (Aulton & Taylor, 2018).
Formula includes solid dispersion, MCC, lactose, starch, talc, and magnesium stearate.
3.2.8 Pre-Compression Evaluation
Parameters included:
These tests follow USP and pharmacopoeial guidelines (USP, 2018).
3.2.9 Post-Compression Evaluation
Tablet properties assessed were:
4. RESULTS AND DISCUSSION
The results obtained from the formulation and evaluation of Ivermectin solid dispersion–based dispersible tablets are presented and discussed in this section. Each experimental step from solubility enhancement to tablet formulation and stability testing has been analyzed in the context of existing scientific literature.
4.1 Solubility Studies
Solubility enhancement was the primary objective of preparing solid dispersions. The solubility of pure Ivermectin was found to be 3.77 µg/mL, confirming extremely low aqueous solubility consistent with reported values (Sweetman, 2017).
After forming solid dispersions with PEG-6000 in different ratios, a substantial increase in solubility was observed. The 1:3 (drug:carrier) ratio demonstrated the highest solubility 266.10 µg/mL representing nearly a 70-fold increase over the pure drug.
This improvement aligns with the mechanisms described by Serajuddin (1999), suggesting increased solubility due to:
These findings are also consistent with previous studies where PEG-based solid dispersions significantly improved the solubility of hydrophobic drugs (Khan et al., 2016; Vasconcelos et al., 2007).
4.2 FTIR Analysis
Fourier Transform Infrared Spectroscopy (FTIR) was performed to determine any drug–polymer interactions affecting chemical stability. Pure Ivermectin exhibited characteristic peaks corresponding to:
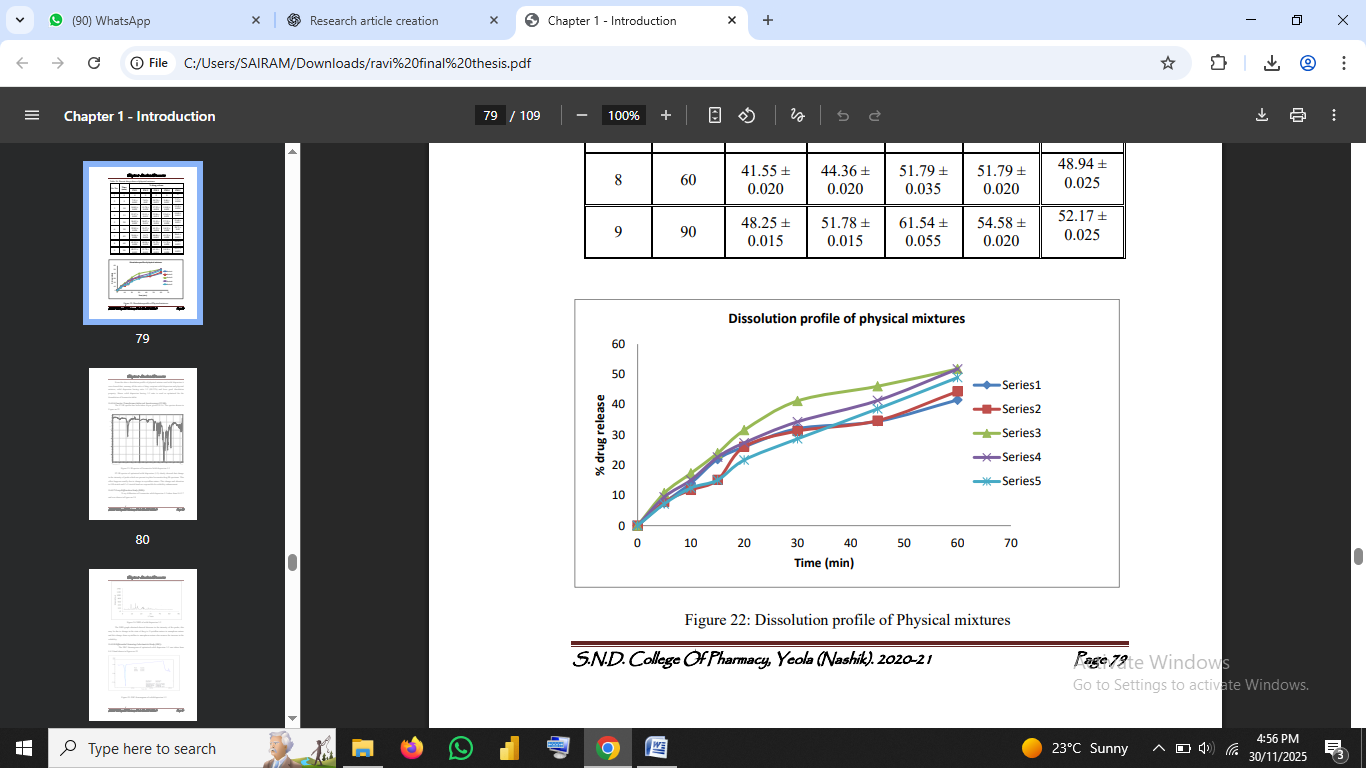
Fig no.5: FTIR spectrum of pure drug vs. optimized solid dispersion
Thus, the polymer does not chemically interact with the drug, affirming formulation stability (Sekiguchi & Obi, 1961)
In the optimized solid dispersion (1:3 ratio), all major functional group peaks of Ivermectin were preserved, with minor peak broadening and intensity reduction. According to Jain et al. (2012), such changes indicate reduced crystallinity or improved hydrogen bonding but do not suggest chemical incompatibility.
4.3 Differential Scanning Calorimetry (DSC)
The slight shift and broadening of peaks indicate a reduction in crystallinity, confirming drug amorphization (Chiou & Riegelman, 1971; Serajuddin, 1999). This supports the observed solubility enhancement, as amorphous drugs typically possess higher free energy and solubility.
4.4 X-ray Diffraction Analysis (XRD)
XRD diffractograms provide direct evidence of crystalline-to-amorphous transformation. Pure Ivermectin showed sharp and intense diffraction peaks, characteristic of crystalline substances.
In contrast, the solid dispersion (1:3) revealed significantly reduced peak intensities, indicating partial or complete amorphization.
The decrease in crystallinity correlates with enhanced solubility and dissolution (Vasconcelos et al., 2007; Khan et al., 2016).
4.5 Scanning Electron Microscopy (SEM)
4.6 Tablet Formulation and Evaluation (F1–F4)
Dispersible tablets were formulated using direct compression a method known for its simplicity and uniformity (Aulton & Taylor, 2018).
Four formulations (F1–F4) were developed by varying excipient proportions. The optimized solid dispersion (1:3) was kept constant across all formulations.
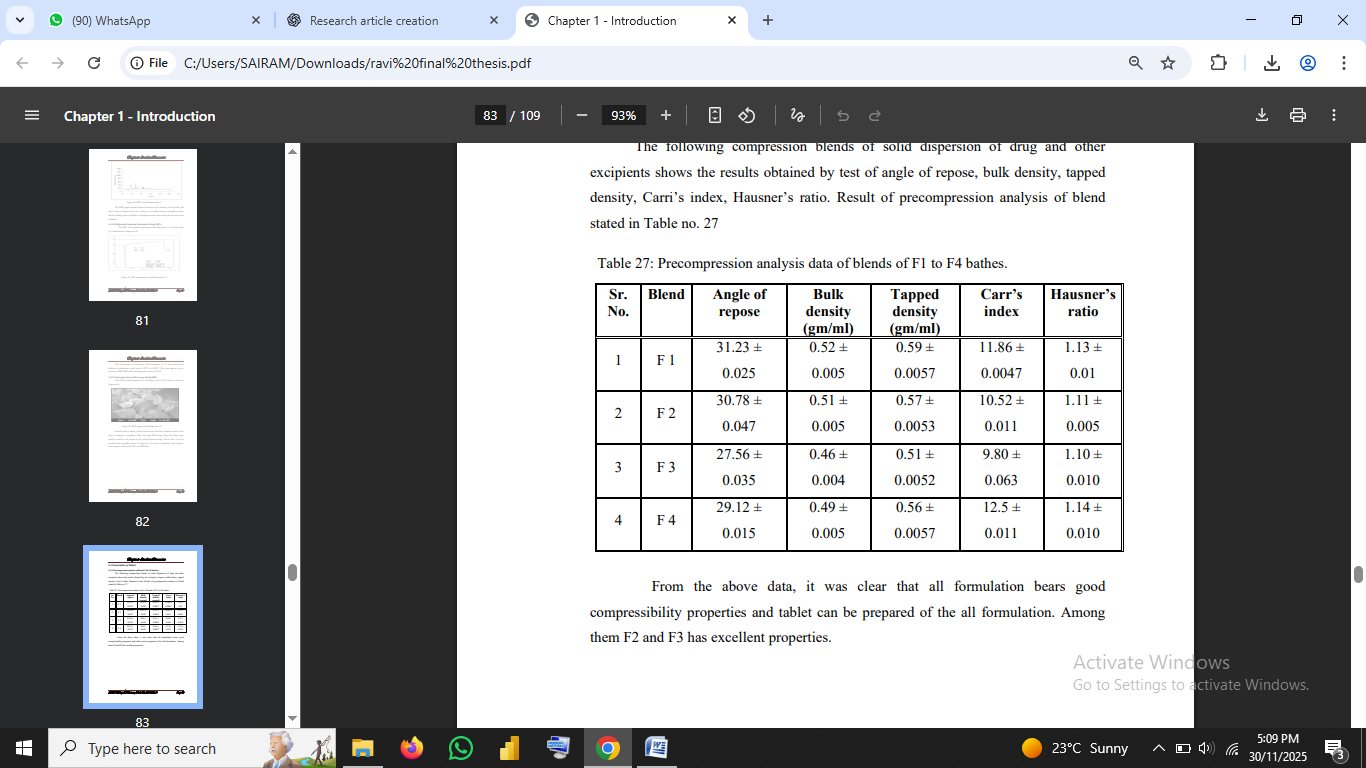
4.7 Pre-Compression Parameters
Pre-compression flow properties indicated excellent compressibility and flow in F3, with the following:
These values fall within the acceptable range, indicating excellent flow (USP, 2018).
Literature suggests that proper flow ensures uniform die filling, reducing tablet weight variation (Aulton & Taylor, 2018).Table no 1: Precompression analysis data of blends of F1 to F4 bathes
4.8 Post-Compression Parameters
4.8.1 Hardness & Friability
F3 tablets exhibited ideal hardness (4.3 kg/cm²) suitable for dispersible tablets.
Friability (0.554%) was well below 1%, satisfying pharmacopeial standards (USP, 2018).
4.8.2 Weight Variation & Thickness
All formulations met acceptable limits, indicating good flow and compression uniformity.
4.8.3 Wetting Time
F3 exhibited the shortest wetting time 19.43 seconds indicating its high hydrophilicity due to PEG and MCC.
4.8.4 Disintegration Time
F3 disintegrated in 18 seconds, far below USP limit of 3 minutes for dispersible tablets.
These findings reflect the formulation’s suitability for pediatric and geriatric populations (Ike et al., 2019).
4.9 In-Vitro Dissolution Studies
Dissolution was assessed using USP Type II apparatus at 50 rpm in 0.1 N HCl.
Formulation F3 demonstrated the highest cumulative drug release (89.74% at 90 min).
The dissolution profile follows Noyes–Whitney principles, where increased wettability and amorphization promote faster drug release (Aulton & Taylor, 2018).
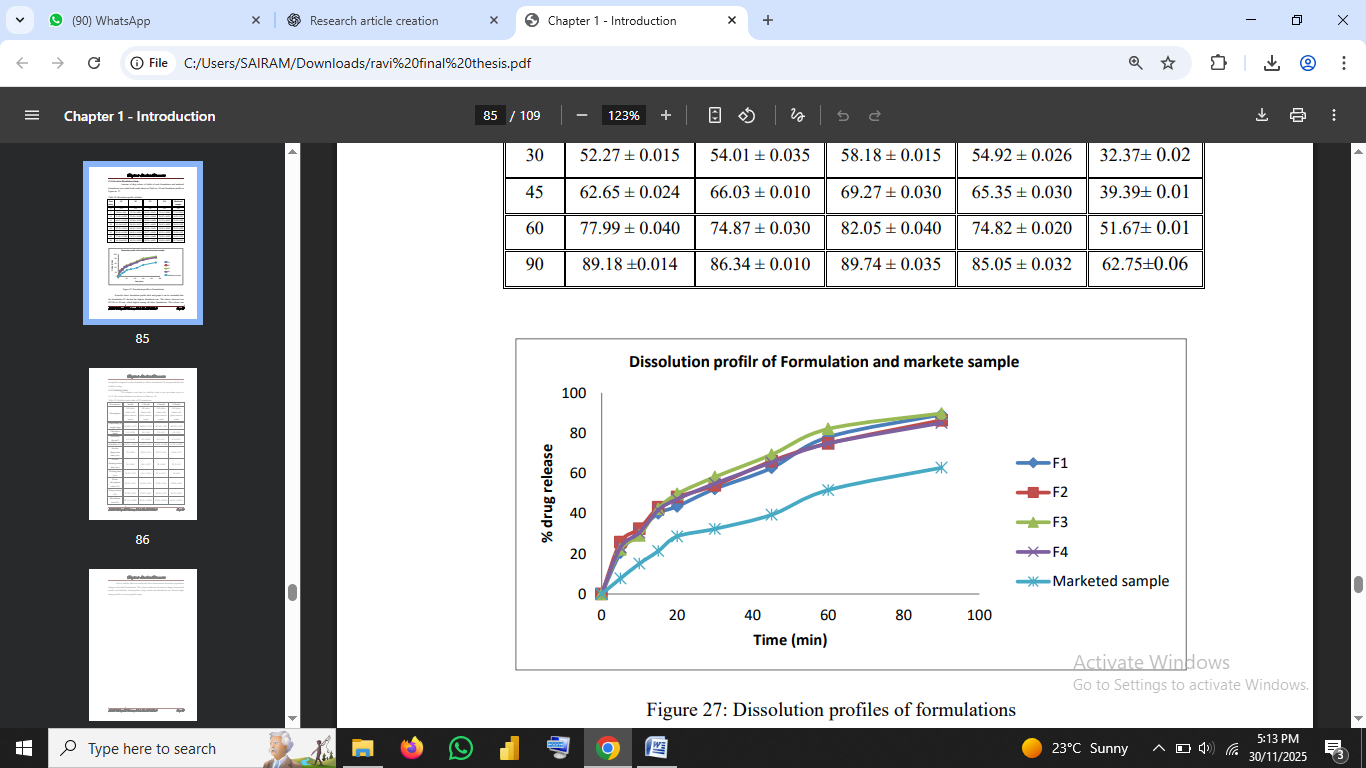
Fig no.6: Dissolution profiles of formulations/Dissolution curve of F1–F4
Scientific Explanation:
PEG-6000 enhances dissolution by:
4.10 Stability Studies
Stability testing was performed under ICH accelerated conditions:
40°C ± 2°C / 75% RH ± 5% for 90 days (ICH, 2003).
Table no.2: Stability study results before and after 90 days
|
Parameters |
Initial |
1 Month |
2 Month |
3 Month |
|
Description |
Off white tablet with plain concave round |
Off white tablet with plain concave round |
Off white tablet with plain concave round |
Off white tablet with plain concave round |
|
Uniformity of weight (mg) |
249.86 ± 0.79 |
248.92 ± 0.76 |
247.56 ± 093 |
246.88 ± 0.87 |
|
Thickness (mm) |
4.5 ± 0.00 |
4.5 ± 0.0 |
4.5 ± 0.0 |
4.5 ± 0.0 |
|
Hardness (kg/cm2 ) |
4.3 ± 0.10 |
4.2 ± 0.05 |
4.5 ± 0.1 |
4.5 ± 0.11 |
|
Friability (%) |
0.554 ± 0.012 |
0.657 ± 0.02 |
0.688 ± 0.005 |
0.729 ± 0.007 |
|
In-vitro dispersion time (sec) |
19 ± 0.00 |
20.33 ± 0.5 |
24.33 ± 0.4 |
24.66 ± 0.2 |
|
In-vitro disintegration time (sec) |
18 ± 0.63 |
18.6 ± 0.57 |
20 ± 0.64 |
22.5 ± 0.5 |
|
Wetting time (sec) |
18.33 ± 0.52 |
21.6 ± 0.15 |
23.3 ± 0.57 |
24 ±0.0 |
|
Water absorption ratio (%) |
63.34 ± 0.51 |
61.69 ± 0.12 |
58.58 ± 0.61 |
59.04 ± 0.49 |
|
Drug content (%) |
97.89 ± 0.23 |
97.07 ± 0.87 |
95.54 ± 0.32 |
95.13 ± 0.07 |
|
Dissolution rate (% |
89.74 ± 0.035 |
86.02 ± 0.026 |
85.06 ± 0.025 |
84.61 ± 0.015 |
Results after 90 days:
These observations match the findings of similar PEG-based formulations reported in stability studies (Singh et al., 2016).
5. CONCLUSION:
The present research successfully achieved its primary objective of enhancing the aqueous solubility and dissolution characteristics of Ivermectin, a BCS Class II drug with an intrinsically low solubility profile. The use of PEG-6000 as a hydrophilic carrier in the preparation of solid dispersions via the fusion method significantly improved Ivermectin’s solubility from 3.77 µg/mL for pure drug to 266.10 µg/mL for the optimized 1:3 solid dispersion, representing a nearly 70-fold enhancement. This substantial improvement aligns with established scientific evidence confirming the efficiency of PEG polymers in enhancing the solubility and wettability of hydrophobic drug molecules (Serajuddin, 1999; Khan et al., 2016).
Characterization using FTIR, DSC, XRD, and SEM revealed that the drug was molecularly dispersed within the PEG-6000 matrix, reducing its crystalline nature and improving its dissolution behavior. FTIR results indicated the absence of chemical interaction between the drug and polymer, ensuring formulation compatibility (Jain et al., 2012). The DSC and XRD analyses clearly demonstrated partial to complete amorphization another contributor to improved dissolution. SEM images further supported this by showing the morphological shift from crystalline needle-shaped particles to smooth, amorphous surfaces.
The optimized solid dispersion was successfully incorporated into dispersible tablet formulations, designed for enhanced patient compliance, particularly among pediatric, geriatric, and dysphagic patients (Ike et al., 2019). Among the four formulations (F1–F4), F3 emerged as the optimized tablet formulation with excellent.
Physicochemical properties:
These findings are consistent with pharmacopeial expectations for dispersible tablets (USP, 2018) and align with principles of rapid dispersion due to hydrophilic excipients (Aulton & Taylor, 2018).
Stability studies conducted under ICH accelerated conditions showed no significant deterioration in physicochemical parameters, confirming the formulation’s robustness and shelf-life stability over 90 days (ICH, 2003). This validates that PEG-6000–based dispersible tablets of Ivermectin remain stable under stress conditions of temperature and humidity.
Overall, the present research demonstrates that solid dispersion using PEG-6000 is a simple, efficient, and scalable formulation strategy for improving the solubility, dissolution, and therapeutic performance of poorly soluble drugs such as Ivermectin. Furthermore, converting the optimized dispersion into dispersible tablets provides an accessible, patient-friendly dosage form with rapid onset of action and better compliance.
Based on the results, the formulation approach used in this study can serve as a model for enhancing the solubility and bioavailability of other hydrophobic, BCS Class II drugs. Future work may explore:
Thus, the study not only contributes to Ivermectin formulation development but also enriches broader pharmaceutical research aimed at improving drug solubility and patient accessibility.
REFERENCES


Yasmin Shaik*, Shilpa Gangurde, Irshad Ahamad, Formulation and Evaluation of Solid Dispersion Based Dispersible Tablets of Ivermectin, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 24-38 https://doi.org/10.5281/zenodo.17774413
 10.5281/zenodo.17774413
10.5281/zenodo.17774413
