Ashokrao Mane College of pharmacy, Peth Vadgaon / Shivaji University, Maharashtra, India 416112
Recently, deep generative models (DGMs) have become revolutionary tools in de novo drug design, allowing for the quick exploration of large chemical spaces that are not possible with conventional techniques. In contrast to traditional high-throughput screening methods, DGMs like reinforcement learning-based frameworks, generative adversarial networks (GANs), and variational autoencoders (VAEs) can create novel molecular structures with desired pharmacological characteristics in silico. These models are able to capture intricate structure–activity relationships and produce candidate molecules with optimal drug-like properties, such as solubility, bioavailability, and target specificity, by utilizing extensive chemical and biological datasets. Advances in multimodal generative approaches, which integrate textual, biological, and chemical data, further improve the ability to suggest compounds with therapeutic relevance and structural novelty. Modern developments have also included human-in-the-loop systems, in which medicinal chemists direct the generative process to guarantee synthetic accessibility and practical viability. In order to improve the interpretability of model decisions and promote trust and adoption in pharmaceutical research, explainable AI techniques are also being developed. Applications show accelerated lead discovery pipelines and span a variety of therapeutic areas, such as neurological disorders, oncology, and antibiotic resistance. Notwithstanding these developments, problems with data quality, model generalization, and establishing a connection between in silico predictions and experimental validation still exist. Future studies will focus on integrating multi-objective optimization, physics-based simulations, and personalized medicine frameworks. All things considered, DGMs have a great deal of potential to transform contemporary drug discovery, lower expenses, and speed up the discovery of innovative treatments for complicated illnesses.
Over the past few decades, drug discovery has largely followed a traditional pipeline involving identification of targets, screening of existing compound libraries (e.g. high?throughput screening), lead optimization through medicinal chemistry, followed by preclinical and clinical trials. This route is time?consuming, expensive, and often limited by the compounds already known or available. Recent advances in artificial intelligence (AI), especially deep generative models, are beginning to shift this paradigm. These models allow for de novo generation of molecular structures (i.e. designing novel molecules from scratch), enabling exploration beyond known chemical space and known scaffolds, potentially accelerating hit identification and lead optimization [1,2].
One of the motivations for using generative AI in de novo design is the sheer size of “chemical space” (i.e. all possible small-molecule compounds). For drug?like molecules, estimates often put this number around 10?? or more, a number utterly beyond what could be fully enumerated or tested experimentally [3]. This vastness implies that existing chemical libraries represent only a tiny fraction of what is possible. Exploring more of this space increases the chance of finding novel bioactive scaffolds with better properties (potency, selectivity, pharmacokinetics, toxicity, etc.). Deep generative models are being used to sample from this enormous space, often guided by property objectives (e.g. binding affinity, drug?likeness, synthetic accessibility) [4,5].
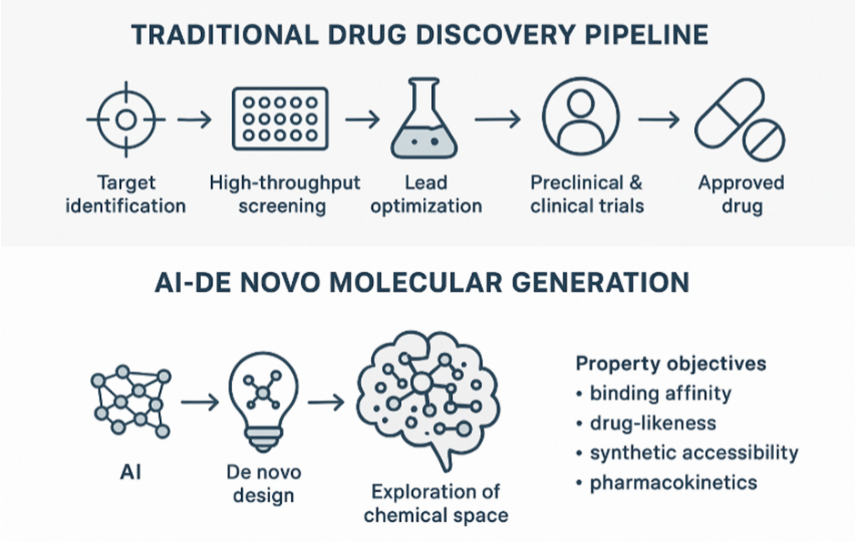
Fig:1 Traditional Drug Discovery Pipeline vs. AI-Driven De Novo Molecular Generation
Deep generative models, such as variational autoencoders (VAEs) and graph neural networks (GNNs), have been instrumental in generating novel drug-like scaffolds. These models learn from existing chemical structures to propose new scaffolds with desired properties. For instance, a study demonstrated the use of a VAE to generate novel scaffolds that adhere to Lipinski's Rule of Five, indicating good oral bioavailability [6].
The optimization of generated molecules to meet drug-likeness criteria is crucial. Deep generative models can be trained to maximize the quantitative estimate of drug-likeness (QED) score, which predicts the likelihood of a compound being orally bioavailable. A study utilizing a conditional graph generative model achieved high QED scores in generated compounds, suggesting their potential as drug-like molecules [7].
Deep generative models facilitate multi-parameter optimization by simultaneously considering various drug-like properties. For example, a study applied a deep learning model to optimize compounds for binding affinity, solubility, and toxicity, achieving compounds that met all desired criteria [8].
Scaffold hopping involves generating new chemical entities by modifying the core structure of known active compounds. A graph diffusion model, DiffHopp, was developed to perform scaffold hopping by generating novel scaffolds that maintain the biological activity of the original compounds [9]. Additionally, a 3D pocket-aware lead optimization model demonstrated the ability to enhance binding affinity and other properties of hit compounds through deep learning-based design [10].
Deep generative models have also been applied to the design of novel bioactive peptides and proteins. For instance, the PepHAR model utilizes hotspot-driven autoregressive generation to design peptides with high binding affinity to target proteins [11]. Furthermore, the DeepTarget model generates novel molecules based solely on the amino acid sequence of the target protein, reducing reliance on prior knowledge [12].
3. Integration with Other Technologies in De Novo Drug Design
a. Coupling Generative Models with Molecular Docking & Molecular Dynamics
Deep generative models have been integrated with molecular docking and molecular dynamics simulations to enhance the design of drug-like molecules with desired binding affinities and stability. For instance, a study by D’Hondt et al. (2025) demonstrated the use of generative adversarial networks (GANs) to generate molecules with high binding affinity to the HIV-1 gp120 protein. These molecules were then subjected to molecular docking simulations using QuickVina 2, followed by MD simulations to assess their stability and interactions within the protein binding pocket. The integration of these techniques facilitated the design of novel inhibitors with promising therapeutic potential [13].
Furthermore, generative models have been employed to learn molecular dynamics trajectories, providing a data-driven approach to simulate and predict the behavior of molecules in complex environments. This approach allows for the generation of flexible, multi-task surrogate models of MD, which can be utilized to predict molecular interactions and optimize drug candidates more efficiently [14].
b. Synergy with Reinforcement Learning for Goal-Directed Molecule Design
Reinforcement learning has been effectively combined with deep generative models to guide the design of molecules towards specific properties, such as high binding affinity or low toxicity. For example, the ACEGEN toolkit, developed by Acellera Therapeutics, integrates RL with generative models to optimize molecular properties by learning from feedback during the design process. This synergy enables the generation of novel compounds that meet predefined criteria, accelerating the drug discovery process [15].
Moreover, a study by Korshunova et al. (2022) highlighted the use of deep reinforcement learning in conjunction with generative models to design inhibitors targeting the epidermal growth factor receptor (EGFR). The approach demonstrated the capability to generate potent inhibitors, which were experimentally validated, showcasing the effectiveness of combining RL with generative modeling in drug design[16].
c. Integration with ADMET Prediction Platforms
The integration of deep generative models with ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) prediction platforms is crucial for designing drug candidates with favorable pharmacokinetic and safety profiles. ADMET-AI, a machine learning platform developed by Swanson et al. (2024), provides fast and accurate ADMET predictions, which can be utilized to evaluate the drug-likeness of molecules generated by DGMs. By incorporating ADMET predictions into the generative design process, researchers can filter out compounds with undesirable properties early in the development pipeline, thereby enhancing the efficiency of drug discovery[17].
Additionally, the incorporation of predictive ADMET models within AI-driven generative chemistry frameworks enables the design of molecules with improved pharmacokinetic properties, as discussed by Zeng et al.[18]. This integration facilitates the generation of bioactive and synthesizable molecules in a time- and cost-effective manner, aligning with the goals of modern drug discovery [19].
4. Case Studies & Breakthroughs
a. In-silico Medicine: From AI Discovery to Clinical Trials
Insilico Medicine has pioneered the use of generative AI in drug discovery, achieving significant milestones. Their platform, Pharma.AI, facilitated the rapid design of ISM001_055, a novel small molecule for idiopathic pulmonary fibrosis (IPF). Remarkably, this candidate progressed from target identification to Phase I clinical trials in just 30 months.[20] Further validating their approach, ISM001_055 entered Phase II clinical trials in 2023, marking the first AI-designed drug to reach this stage [21]. Additionally, the U.S. Adopted Names Council officially named Rentosertib, an AI-designed drug for IPF, underscoring the growing recognition of AI's role in drug development [22].
b. BenevolentAI: Advancing AI-Designed Therapeutics
BenevolentAI has leveraged AI to accelerate drug discovery, with several candidates progressing through clinical trials. In 2024, the company announced positive safety and pharmacokinetic data from a Phase Ia clinical study of BEN-8744 in healthy volunteers, indicating the potential of AI-designed drugs to meet safety benchmarks.[23] Furthermore, their collaboration with the Drugs for Neglected Diseases initiative (DNDi) aims to identify potential biological targets and drug repurposing candidates for diseases like dengue, highlighting AI's versatility in addressing diverse therapeutic areas [24].
c. AI in Targeting Kinases, GPCRs, and Viral Infections
AI has been instrumental in designing inhibitors targeting kinases and G-protein-coupled receptors (GPCRs), which are pivotal in various diseases. For instance, the Kinase drUgs mAchine Learning framework (KUALA) utilizes AI to identify kinase-active ligands, facilitating drug repurposing and novel inhibitor development [25,26] Moreover, AI-driven virtual screening has been employed to discover small molecules that inhibit the S-ACE2 interaction, a critical step for SARS-CoV-2 entry, showcasing AI's role in combating viral infections [27,28].
5. Advantages Over Traditional Methods
Deep generative models significantly accelerate the identification of potential drug candidates by rapidly generating novel molecular structures. This speed is achieved through the models' ability to learn complex patterns from large chemical datasets, enabling the generation of bioactive molecules in a fraction of the time compared to conventional high-throughput screening methods. For instance, studies have demonstrated that DGMs can produce viable drug-like molecules more swiftly than traditional approaches, thereby reducing the time required for hit identification [29].
DGMs excel in exploring the vast and complex chemical space by generating diverse molecular structures that adhere to desired properties. Unlike traditional methods that may rely on predefined libraries, DGMs can sample from a broader range of chemical entities, uncovering novel compounds with potential therapeutic effects. Research indicates that DGMs can effectively navigate chemical space, identifying molecules that traditional methods might overlook [30].
By utilizing computational models to predict molecular properties and interactions, DGMs minimize the need for extensive laboratory experiments, leading to significant cost savings. This reduction in experimental workload not only cuts down on financial expenditures but also decreases the time spent on empirical testing. Studies have highlighted that DGMs contribute to cost efficiency and reduce the experimental testing required in drug discovery [31].
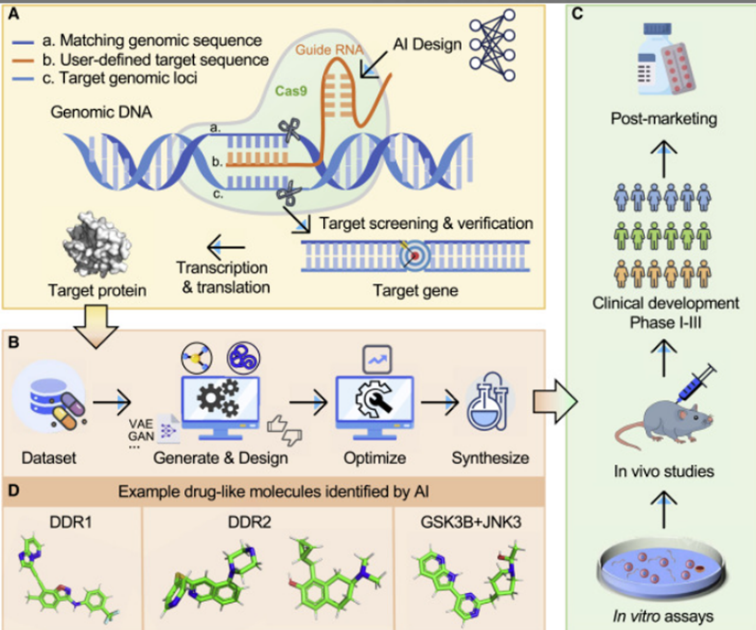
Fig:2 "AI-Assisted Drug Discovery Workflow: From Target Identification to Clinical Development"
5. Challenges and Limitations
a. Synthetic Feasibility of AI-Generated Molecules
While deep generative models can design novel molecules, many of these compounds may be synthetically infeasible. Traditional synthetic accessibility scores often fail to predict the practical challenges associated with these molecules. To address this, the Retro-Score (RScore) has been introduced, which computes a synthetic accessibility score by performing a full retrosynthetic analysis through Spaya, a data-driven synthetic planning software. This approach highlights the importance of conducting comprehensive retrosynthetic analyses to determine the synthesizability of AI-generated molecules [32].
Furthermore, machine learning tools are increasingly being utilized to predict synthetic feasibility early in the drug development process. These tools help drug developers avoid costly failures, streamline research and development, and design molecules that are both effective and practical to produce [33].
b. Model Interpretability and Transparency
The "black-box" nature of deep generative models poses significant challenges in understanding how these models arrive at specific predictions. This lack of transparency complicates the validation and trustworthiness of AI-generated drug candidates. To mitigate this issue, explainable artificial intelligence (XAI) approaches are being employed to make models more interpretable. For instance, perturbing the input or parameters in the model and observing how the results change can help in understanding the model's decision-making process [34].
The efficacy of deep generative models is heavily dependent on the quality and diversity of the data they are trained on. Training models on unrepresentative datasets can lead to overfitting and the generation of biased or unrealistic outputs. Moreover, experimental data in drug discovery is often sparse, noisy, and lacks standardization, making it challenging to integrate and analyze at scale [35].
To address these issues, efforts are being made to improve data standardization and foster collaboration between AI researchers and pharmaceutical experts. These initiatives aim to enhance the quality of data and ensure that AI models are trained on diverse and representative datasets [36].
6. Regulatory and Ethical Concerns in AI-Designed Drugs
The integration of AI in drug development raises several regulatory and ethical challenges. Regulatory bodies face difficulties in overseeing AI/ML medical devices due to their evolving nature and the complexity of AI algorithms. For example, the FDA has authorized numerous AI/ML-enabled medical devices but struggles with regulating them effectively [37].
Ethically, the use of AI in drug development can lead to concerns about patient data privacy, accountability, and informed consent. There is also the risk of perpetuating biases if AI models are trained on non-diverse datasets, which can affect clinical outcomes. Therefore, establishing clear ethical frameworks and regulatory guidelines is crucial to ensure the responsible use of AI in drug discovery [38].
a. Multimodal Generative Models in Drug Design
Recent advancements have led to the development of multimodal generative models that integrate various data types—such as text, chemical structures, and biological information—to enhance drug discovery processes. For instance, Chem3DLLM utilizes large language models to generate molecular structures by processing both textual and chemical data, facilitating the design of novel compounds with desired properties. Additionally, Token-Mol employs a token-based approach to encode 2D and 3D structural information, along with molecular properties, into discrete tokens, enabling the generation of drug-like molecules with complex architectures. These multimodal systems aim to bridge the gap between different data types, offering a more holistic approach to drug design. [39].
b. Human-in-the-Loop Drug Design Systems
Integrating human expertise into AI-driven drug design processes has proven beneficial in refining molecular design strategies. Human-in-the-loop systems combine machine learning models with chemist input to adapt scoring functions, ensuring that generated compounds align with specific design goals. This approach enhances the adaptability and accuracy of drug design, especially in complex scenarios where human judgment is crucial. For example, such systems have been applied to optimize compound libraries by incorporating expert feedback into multi-parameter optimization frameworks. [40].
c. Explainable AI (XAI) in Rational Drug Design
As AI models become more complex, ensuring their decisions are interpretable is vital for their acceptance in regulated industries like pharmaceuticals. Explainable AI (XAI) frameworks have been developed to elucidate the reasoning behind AI-generated predictions, such as drug-target interactions and molecular properties. These frameworks enhance the transparency of AI systems, allowing researchers to understand and trust the model's outputs, which is essential for regulatory approval and clinical application. [41].
d. Expansion Toward Personalized and Precision Medicine
Generative AI is increasingly being utilized to tailor drug discovery processes to individual patient profiles, advancing the field of personalized medicine. By modeling disease progression and predicting patient-specific drug responses, AI systems can identify optimal therapeutic strategies for individuals. This personalized approach not only improves treatment efficacy but also reduces adverse effects by aligning therapies with the unique genetic and phenotypic characteristics of patients. The integration of generative AI into personalized medicine holds promise for more effective and targeted treatments.[42].
CONCLUSION
The limitations of conventional discovery pipelines, which mainly rely on known chemical libraries and time-consuming screening, are addressed by deep generative models (DGMs), which represent a revolutionary step in de novo drug design. DGMs facilitate effective navigation of the vast chemical space by utilizing generative adversarial networks, variational autoencoders, and reinforcement learning to produce novel molecular structures with optimal physicochemical and pharmacological profiles. In addition to speeding up the initial phases of drug discovery, these models enable multi-objective optimization, striking a balance between synthetic feasibility and drug-likeness, bioavailability, and target specificity.
The integration of multimodal generative frameworks—combining chemical, biological, and textual data—enhances predictive accuracy and therapeutic relevance, while human-in-the-loop systems ensure practical alignment with medicinal chemistry principles. Furthermore, the growing emphasis on explainable AI fosters trust, interpretability, and broader adoption in pharmaceutical research. Despite these advances, challenges remain, particularly regarding data quality, model generalization, and bridging the gap between in silico predictions and experimental validation. Looking forward, the integration of physics-informed simulations, robust multi-objective optimization, and personalized medicine frameworks will further strengthen the impact of DGMs. Ultimately, these innovations hold the potential to significantly reduce drug development timelines, costs, and risks, ushering in a new era of precision-driven, AI-enabled therapeutics.
REFERENCES


Pradnya Ghatage, Tejashree Khamkar, Rutuja Chougule, Tanuja Chogule, Rutuja More, Emerging Application Progress of Deep Generative Models in De Novo Drug Design, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 10, 752-761. https://doi.org/10.5281/zenodo.17295131
 10.5281/zenodo.17295131
10.5281/zenodo.17295131
