Dr. L H Hiranandani College of Pharmacy, Ulhasnagar, Maharashtra, India
The present study aimed to develop and evaluate a suitable oral dosage form of Levodopa for the effective management of Parkinson’s disease. An Orally Disintegrating Tablet (ODT) of Levodopa was developed using co-processed excipient F-Melt Type C as a filler-disintegrant base, Crospovidone as a superdisintegrant, and Sodium Stearyl Fumarate as a lubricant. A 2³ factorial design was employed to optimize the formulation, considering concentrations of F-Melt, Crospovidone, and Sodium Stearyl Fumarate as independent variables, while tablet hardness, disintegration time, and drug release were selected as dependent variables. Pre-compression parameters including bulk density, tapped density, angle of repose, Carr’s index, and Hausner’s ratio confirmed good flowability of powder blends. Post-compression evaluation revealed acceptable limits for friability, hardness, weight variation, disintegration time, drug content uniformity and wetting time. Studies on in vitro dissolution demonstrated rapid and complete drug release in (SSF) simulated salivary fluid and phosphate buffer. The batch that was optimized showed a DT time of less than 30 seconds with >90% drug release within 15 minutes, ensuring enhanced patient compliance. Stability studies conducted under ICH guidelines confirmed the robustness of the optimized formulation. The study successfully established Levodopa ODT as a patient-friendly dosage form with potential to improve therapeutic results in Parkinson’s disease.
Parkinson's disease is defined pathologically by dopaminergic neuron degeneration in the substantia nigra of the midbrain, which causes pathophysiologic abnormalities in the neural networks of the downstream basal ganglia. This is a multifactorial degenerative disease of the central nervous system that progresses over time. The clinical manifestations of (PD) Parkinson's disease frequently arise from the progressive loss of dopaminergic nigrostriatal neurons, resulting in dopamine deficit that governs movement and balance.The disease is characterized by bradykinesia, slowness of movement, and at least one other motor feature such as rigidity or rest tremor. Patients suffering from Parkinson’s disease frequently exhibit non-motor symptoms. The loss of dopaminergic cells in brain leads in motor defects, with about 60-70% of neuronal cell loss occurring at the time of diagnosis [1]. Dopamine replacement with oral levodopa still remains the leading standard for symptomatic therapy. To date, the absence of techniques for the early detection of Parkinson’s Disease is a major hurdle in the path of PD treatment. Low oral bioavailability (30%) [2], peripheral decarboxylation of enzymes by dopa decarboxylase in the kidney, gut and liver and low brain uptake, diminish the quantity of levodopa accessible for brain absorption are all obstacles to levodopa's effectiveness [3]. Levodopa combined with a (DCI), Dopa decarboxylase inhibitor together such as Benserazide or Carbidopa, is regarded the ultimate first-line treatment for Parkinson’s Disease. DCIs prevent levodopa from peripherally converting to dopamine before it passes through the (BBB)blood-brain barrier [4]. Oral drug administration is widely accepted, accounting for 50–60% of all dosage forms. Solid dosage forms are especially popular due to their ease of use, accurate dosing, suitability for self-medication, non-invasiveness, and high patient compliance. Both locally acting and systemic acting drugs can be administered by this route. The patient population, however, consistently favors the oral method of administration over all other modalities. Despite injectable 100% bioavailability, which gives them an advantage over solid dose forms, solid oral dose forms like capsules and tablets are still preferred by patients. Solid dosage forms offer the important benefits of self-medication, accurate dosing, and non-invasive delivery, relative to injectable that require syringe assistance. Nevertheless, the most popular formulas have certain disadvantages as well [5]. Dysphagia or trouble in swallowing; is the major challenge faced by many patient such as bedridden patients, geriatric, paediatric, nauseated, and psychiatric patients. Overcoming this Orally Disintegrating systems were placed in market [6]. To overcome challenges related with oral route such as extensive hepatic first pass effect and degradation of drug in gastrointestinal tract (GIT). The Orally Disintegrating Tablet (ODT) allows localized levodopa administration, potentially enhancing its targeted absorption. This may improve symptom management in Parkinson's condition by aiding drug transport across the blood-brain barrier. In this research, we have formulated levodopa ODT’s using Co-processed excipients, superdisintegrants such as Cross-povidone to achieve optimal tablet disintegration& lubrication. A 23 Factorial design was also applied for optimisation. For orally disintegrating tablet, due to the limited volume of media present in oral cavity region traditional dissolution techniques might not be directly relevant. To tackle this difficulty, a novel approach was developed to replicate the circumstances of the oral cavity region in order to investigate the disintegration and dissolution of tablets.
MATERIALS AND METHODS
Materials
Levodopa was procured from Molychem Pvt Ltd, Mumbai, India. Ludiflash (co-processed excipient) was given as free gift samples by BASF, India. F-MELT TYPE C (co-processed excipient) was procured as free sample by Fuji Chemical/Barentz India. Crospovidone was super disintegrant. Sodium stearyl fumarate (Lubricant) was procured as free sample from Umedica Laboratories Pvt.Ltd.
Preformulation Studies
In this research, preliminary batches were developed with an aim to find the best suitable ODT excipient. The choice of suitable excipients for the creation of an efficient and stable dosage form is made simpler by these studies. Prepared various batches with different combinations of excipient and evaluated the results. Pre- formulation studies aim to find those physicochemical characteristics and excipients that might affect formulation design, technique of production, and pharmacokinetic biopharmaceutical characteristics of the obtained product.
Melting point
Levodopa’s purity was assessed by measuring its melting point through capillary tube technique. The thin glass capillary tube was filled with a small quantity of drugs, mounted to the thermometer, and then partially submerged in Thiele’s tube filled with liquid paraffin. The sample inside the capillary melted completely when the Thiele tube underwent heating, indicating the drug’s melting temperature.
Solubility of drug in different Solvents
According to the I.P.2018, a study on solubility was conducted. In this case, the volume of the maximum solvent needed for the solute to dissolve was found. The drug's solubility was checked in various solvent like water, phosphate buffer pH 6.8, ethanol, methanol, and (SSF) simulated salivary fluid.
Determination of λ max
A Shimadzu UV-visible spectrophotometer with 10 mm quartz cuvettes was used by the UV system to determine λ max and create a calibration curve. Levodopa was accurately weighed and dissolved in a phosphate buffer pH 6.8 to prepare a 1000 μg/ml stock solution. This stock solution was then exposed to further dilution with the phosphate buffer to achieve a concentration of 100 μg/ml. The drug’s UV spectrum was measured over range of 200-400 nm, using the blank as the same phosphate buffer solution. The levodopa’s λ max was determined by analyzing the absorption peaks within this range. Similar was done using (SSF) simulated salivary fluid of pH 6.8.
Drug- excipients Interaction studies [7]
These studies were conducted by FTIR (Shimadzu IR Spirit, Lab solution, Japan, XRD & DSC (DSC25 TA, Instruments).
Method Of Preparation of Levodopa ODT
ODT’s containing Levodopa were formulated by applying a direct compression technique. All the ingredients Drug Levodopa, F-Melt® Type C, Crospovidone, were screened through a 0.150 mm sieve (No.60) before mixing to ensure uniform particle size distribution. Required quantity of drug, co-processed excipient, super disintegrant were blended in a sealable polythene bag for 20 min. Sodium stearyl fumarate was added as the lubricant to the powder mixture and blended. Compression of the ODT were processed utilizing Rimek Minipress 1 Multistage tablet punching unit equipped with a 9 mm round flat punch set. Thickness of tablets was around 3 mm and each tablet's weight of 200 mg was maintained.
Optimization of Orally Disintegrating Tablet by Using 23 Factorial Design[8]
Factorial design, a statistical optimization method, was used. to obtain an optimized formula for levodopa (ODT)orally disintegrating tablet. A 23 full factorial experimental design approach was adopted, where three factors at two levels of each (23) were identified as experimental design. The quantities of F-Melt® Type C co-processed excipient (X1), super disintegrant Crospovidone (X2) and Sodium stearyl fumarate (X3) were selected as the independent variables in the study. The two levels (-1, +1) of these independent variables were identified from the outcomes obtained in the analysis of initial batches. All other variables, including Drug and environmental conditions, were kept constant throughout the study. Key properties such as (DT)Disintegration time, Hardness & drug release % in SSF & Buffer were evaluated as response variables to identify the optimal formulation. This systematic approach enhances the understanding of how excipients affect tablet performance, ultimately aiming to enhance treatment outcomes for patients.
Composition of Salivary Fluid (SSF) [9]
Each test tube containing five millilitres of distilled water was filled with all the chemicals needed to prepare SSF. All test tube were swirled until the contents were fully dissolved in the distilled water. Then, in the specified order, the components were added to the beaker. If any particles were seen, Whatman filter paper was utilized to filter the mixture, and the obtained solution was used for the given studies.
Table 1. Composition of saliva [9]
|
Sr. no. |
Ingredients |
gm/ 100ml |
|
1 |
Potassium chloride (KCl) |
0.096 |
|
2 |
Sodium Bicarbonate NaHCO3 |
0.063 |
|
3 |
Sodium Chloride (NaCl) |
0..012 |
|
4 |
Potassium Thiocyanate (KSCN) |
0.018 |
|
5 |
Potassium Dihydrogen Phosphate (KH2PO4) |
0.065 |
|
6 |
Urea |
0.020 |
|
7 |
Sodium Sulfate (Na2SO4) |
0.076 |
|
8 |
Ammonium Chloride (NH4Cl) |
0.017 |
|
9 |
Calcium Chloride (CaCl2) |
0.022 |
Evaluation of precompression powder blend
The blend's flow characteristics, such as the Hausner's ratio, Carr's Index, and angle of repose, fall within the expected range for tablet formulation. The flow characteristics of the excipients and the active pharmaceutical ingredient (API) are crucial in selecting the best compression method for tablets [10].
Tan θ = h/r
Bulk Density and Tapped Density
Loose bulk density was determined by pouring a weighed quantity of blend into a measuring cylinder and measuring the volume and weight [11].

The cylinder was tapped against a hard surface at two-second intervals from a height of 10 cm in order to measure tapped bulk density (TBD). The tapping persisted until no more change in volume was seen [11].

Carr’s index
The index of compressibility of the blends was calculated by Carr’s compressibility index. It is determined by the following formula: [11]
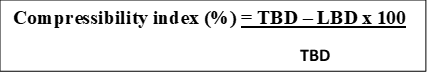
Hausner’s ratio
The measurement of the frictional resistance is Hausner’s ratio. The ideal range should be 1.2-1.5. It is determined by using the following formula: [11]

Post compression Parameter’s Evaluation
Hardness
A tablet's ability to resist chipping, abrasion, or breaking during handling, storage, and transportation is reflected in its hardness, which is described as the force needed to break it throughout its diameter. The hardness ensures that the tablet maintains its integrity before usage, preventing damage. The Monsanto Hardness Tester was utilised to determine the hardness of all tablet formulation, a widely used technique for evaluating tablet strength and durability [12].
Tablet thickness & diameter
Thickness is an important characteristic in reproducing appearance of tablet. Tablet thickness can be determined with an easy technique. Using a digital Vernier caliper, the thickness of five tablets was determined [12].
Weight variation
20 tablets were selected randomly from every batch & randomly weighed. The average weight & standard deviation of 20 tablets were calculated. A batch passes the weight variation test when none of the units differ from the average weight by more than the specified percentage or exceed the limits given in the table [12].
Table2: Limits for % deviation for tablets
|
Average weight of tablet (mg) |
Percentage deviation |
|
80 or less |
10 |
|
80 to 250 |
7.5 |
|
More than 250 |
5 |
Friability test
Friability testing of the tablets was conducted using the Roche friabilator. From each formulation, twenty tablets were picked randomly, their initial weight was determined, and were then put into the friabilator. For four minutes, the friabilator runs at 25 rpm (100 revolutions). After being de-dusted, the tablets were weighed once more (final weight). The following formula was utilized to get the % friability [13].
Where, F= % Friability. Critera is it should not be more than 1%
F = ???????????????????????????? ????????????????????????-???????????????????? ????????????????????????
Initial ???????????????????????? × 100
Uniformity of Drug Content
A random sample of tablets were taken from each prepared formula. They were crushed to obtain fine powder. Powder equivalent to 100 mg of levodopa were accurately weighed and dissolved in a100 ml phosphate buffer into a 100 ml volumetric flask. This was sonicated for 10 mins. Use Whatman filter paper to filter the stock solution. From the above solution, stock solution of 1ml was diluted to 10 ml. Spectrophotometry was utilized to measure the drug content at 280 nm.

In-vitro drug release/Dissolution
Release of drug from Levodopa ODT in vitro was conducted in franz diffusion cell placed on magnetic stirrer. A dissolution medium of total of 20 ml of the simulated salivary fluid (SSF) at pH 6.8, was introduced to the beaker, and the temperature was maintained at 37 ± 0.5°C temp to simulate body conditions. To ensure temperature equilibrium, 2 ml of the sample was withdrawn at pre-planned intervals (0,5,10,15,20,25 & 30) mins, filtered using Whatman filter paper, and with an equal volume of fresh medium was replinshed. The concentration of drug in the samples was found using UV spectroscopy at 280 nm. 3 tablets were examined for each time point, and the total % of drug released was calculated. Similar procedure was used to determine the drug release in phosphate buffer as well.
In-vitro Disintegration Test
Tablet disintegration time was determined by utilizing a beaker containing SSF pH 6.8.and temperature maintained at 37°C ± 0.5°. Tablets from each batch were introduced in a beaker with SSF, and their disintegration time was noted in seconds.
Wetting Time Test
A folded piece of tissue paper (diameter 10cm) was kept in a small petri dish (internal diameter10 cm) containing water with a dye solution. The tablet’s wetting time was determined by placing it on tissue paper in a dish and measuring the time it required for water to reach the tablet's upper surface [14].
Statistical Analysis of Optimization Data
In this study DOE (Design of Experiments software,Stat Ease version 13) was used for statistical data analysis model optimization, Multiple linear regression analysis produced polynomial equations for each dependent variable in the form of y = f(x). Based on comparison of several statistical criteria including adjusted R2, predicted R2 and p value, the best model fitting the data was selected. In order to identify the variables that significantly affected the dependent variables, analysis of variance (ANOVA) was also employed. Response surface graphs such as contour plots and 3D surface plots are utilized to graphically represent the relationship between these independent and dependent variables. These plots can also be used to identify the reaction of dependent variables at the intermediate levels of independent factors, as well as the impact of independent factors at a specific level over the dependent variables. In the end, the desirability approach was used to create an optimum formulation with the desired results utilizing these polynomial equations and response graphs.
Short-term Stability Studies
An analysis of the optimized batch for Short-term stability was conducted following ICH guidelines at 40°C ± 2°C and 75% ± 5% RH. Also, evaluation of optimized batch were done at room temperature. The samples were sealed in pouches and stored in amber-colored bottles to protect them from light and moisture. Stability assessments were conducted at one-month intervals, with evaluations focusing on key tablet properties such as appearance, disintegration time, hardness, drug release profiles. The results provide critical data on the formulation's ability to maintain its quality and effectiveness under stress conditions over a defined period, supporting the product's shelf life.
RESULTS AND DISCUSSION
Melting point
Levodopa exhibited a melting point in the range of 296–298 °C.
Solubility
The solubility of the levodopa drug was evaluated in various solvents. The results indicated that it was sparingly soluble in methanol, slightly soluble in water and ethanol, insoluble in chloroform. However, good solubility was found in Formic acid, Phosphate buffer (pH 6.8) and Simulated salivary fluid (SSF, pH 6.8). These solubility characteristics indicate that it might be effectively formulated for therapeutic administration, as its solubility in biologically relevant media suggests potential for absorption and therapeutic efficacy.
Spectral characterization by UV method
UV spectrum analysis of levodopa was conducted in phosphate buffer pH 6.8 and SSF pH 6.8 [Figures 1&2] shows the spectrum. The drug's UV spectra revealed a maximum absorption at 280 nm.

Figure 1: UV spectra of the Levodopa in phosphate buffer pH 6.8

Figure 2: UV spectra of the Levodopa in SSF pH 6.8.
Calibration curve of Drug in SSF pH 6.8
The curve indicated that levodopa follows Beer’s law and has the linearity range of 10-50μg/ml. The obtained experimental data (Y = 0.0011x + 0.0252) having a correlation coefficient of 0.9973 were utilized to determine the levodopa concentration in the In-vitro dissolution study.
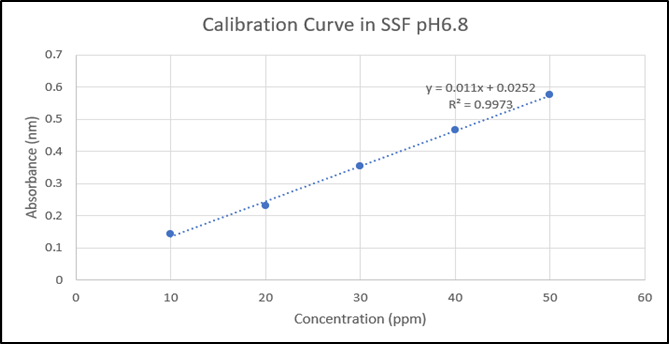
Figure 3: Levodopa Calibration curve in SSF pH 6.8 at 280nm
Calibration curve of Drug in Phosphate Buffer pH 6.8
The curve indicated that levodopa follows Beer’s law and has the linearity range of 20-100μg/ml. The obtained experimental data (Y = 0.0135x + 0.018) having a correlation coefficient of 0.99973 were utilized to determine the levodopa concentration in the In-vitro dissolution study.
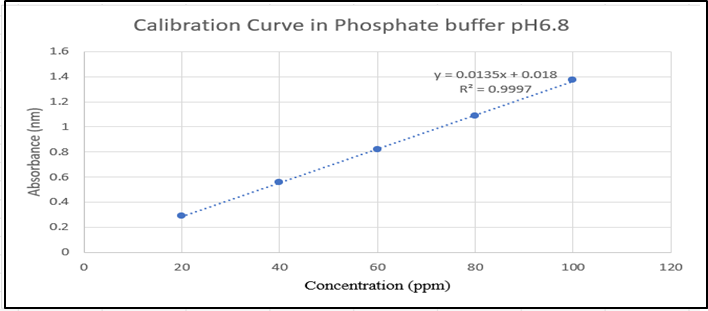
Figure 4: Levodopa Calibration curve in Phosphate buffer pH 6.8 at 280nm
Drug- Excipients Interaction Studies:
Interaction study by UV-spectroscopy
Interaction study by UV-spectroscopy showed no possible interaction between drug &excipient. The overlay plot of Levodopa & F-Melt® Type C does not exhibit any interaction. This indicates the absence of interaction between them.
Figure 5: Spectra of F-Melt® Type C

Figure 6: Spectra of Levodopa
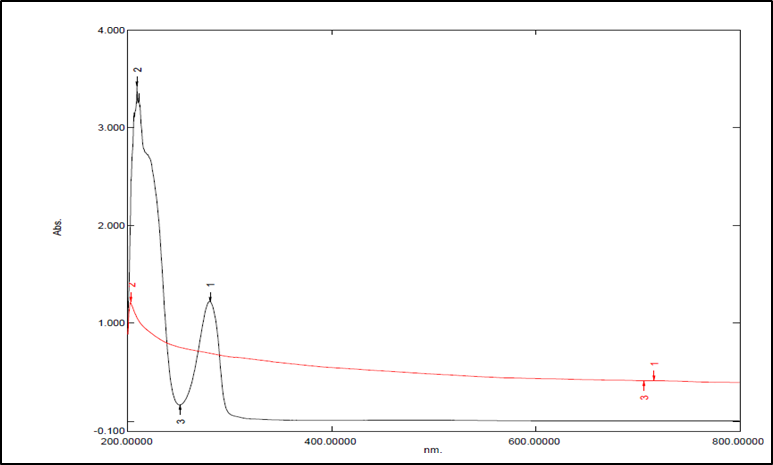
Figure 7: Spectra of Levodopa & F-Melt® Type C
Fourier Transform Infrared (FTIR) spectroscopy studies
The FTIR analysis exhibited no significant differences in peak values between drug and a mixture of excipients and drug, indicating absence of interaction between the two. The FTIR spectra of pure drug Levodopa exhibited distinct bands at the following wavelengths.
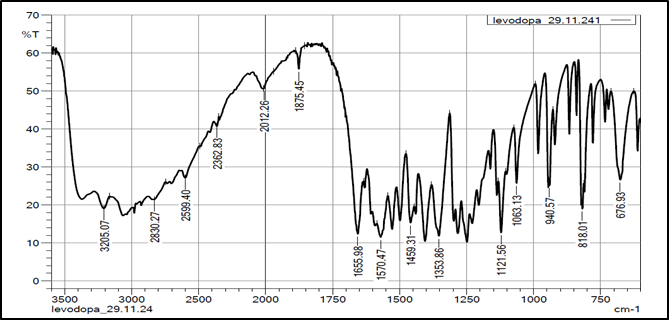
Figure 8: FTIR spectra of Levodopa
Table 3: Results of FTIR.
|
Functional group |
Range (cm−1) |
Obtained value (cm−1) |
|
Amines |
3200-3500 |
3205.07 |
|
Carboxylic acid |
2500-3300 |
2599.40 |
|
-C=O |
1640-1800 |
1655.98 |
|
C-H aromatic ring |
650-1000 |
818.01 |
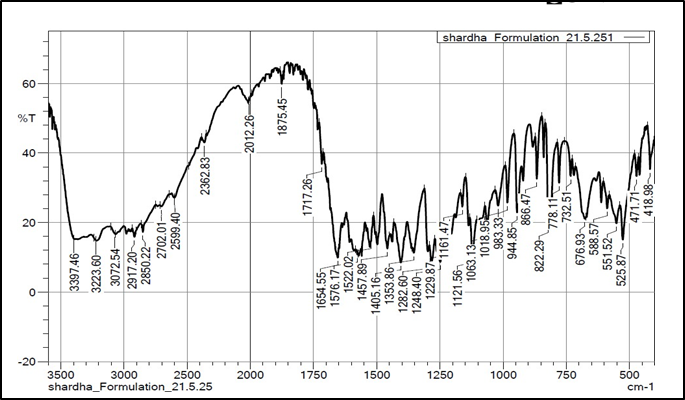
Figure 9: FTIR spectra of Levodopa ODT Formulation
X-Ray Diffraction study (XRD):
XRD of the Levodopa is seen in [Figure 10]. The XRD of Levodopa shows sharp crystalline peaks.
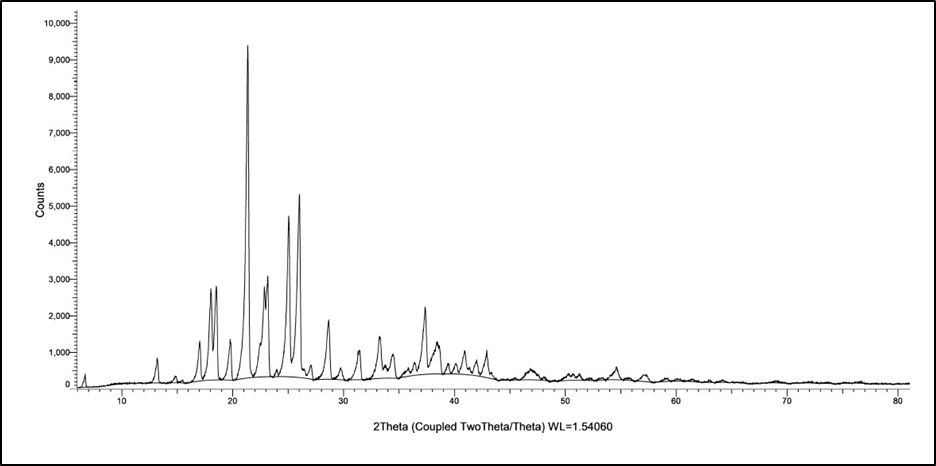
Figure 10: XRD spectra of the Levodopa
DSC
A DSC thermogram profile of pure Levodopa demonstrated a pronounced endothermic peak at 298.55°C. The formulation, had corresponding endothermic peaks at 265.85°C. The shifting in the melting point is associated to change in nature from crystalline to amorphous form. A peak at 166.26 °C is of excipient seen in DSC of formulation. This compatibility suggests that the formulation is stable and retains the drug's integrity, which is important for ensuring optimal therapeutic performance and efficacy in clinical applications.
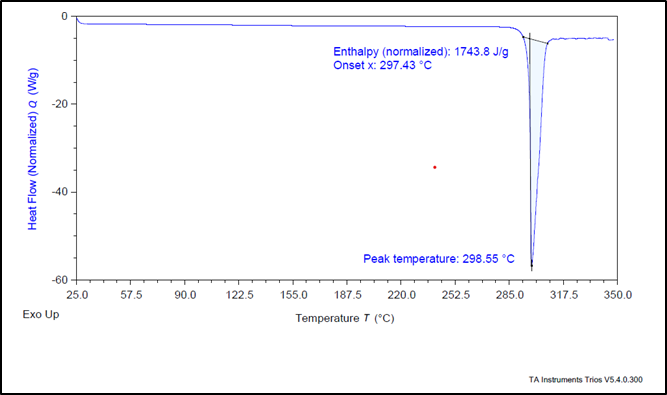
Figure 11: DSC Thermogram of pure Levodopa

Figure 12: DSC Thermogram of Optimized Formulation
Optimization of Levodopa Orally Disintegrating Tablet by Using 23 Factorial Design :
The factorial 23 design was utilized to optimize the amount of co-processed excipient, superdisintegrant &lubricant. F-Melt® Type C, Crospovidone and sodium stearyl fumarate were used as independent variables. The dependent variables included Disintegration Time (sec), Hardness (kg/cm2) % release of drug in Phosphate buffer & SSF. Total 8 formulations were formulated according to the 23factorial design and evaluated. All the eight formulation batches of 23factorial design are compiled in table.
Table 4: The23 full factorial design layout of Levodopa Orally disintegrating tablets
|
Ingredients (in mg) |
Formulation code |
|||||||
|
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
|
|
Drug |
100 |
100 |
100 |
100 |
100 |
100 |
100 |
100 |
|
F-melt Type C |
72 |
86 |
72 |
86 |
86 |
86 |
72 |
72 |
|
Cross povidone |
24 |
10 |
10 |
24 |
24 |
10 |
10 |
24 |
|
Sodium Stearyl fumarate |
2 |
4 |
4 |
4 |
2 |
2 |
2 |
4 |
|
Total (mg) |
198 |
200 |
186 |
214 |
212 |
198 |
184 |
200 |
Table 5: Experimental Batches of 23 factorial design with observed responses
|
Formulation |
Factor 1 A: FMELT Type C (mg) |
Factor 2 B: Cross povidone (mg) |
Factor 3 C: Sodium Stearyl Fumarate (mg) |
Response 1 Disintegration Time (sec) (R1) |
Response 2 Hardness (kg/cm2) (R2) |
Response 3 Drug release in Buffer (%) (R3) |
Response 4 Drug release in SSF (%) (R4) |
|
F1 |
72 |
24 |
2 |
16 |
3.2 |
103 |
105 |
|
F2 |
86 |
10 |
4 |
20 |
3.4 |
100 |
96 |
|
F3 |
72 |
10 |
4 |
19 |
3.2 |
97 |
100 |
|
F4 |
86 |
24 |
4 |
13 |
3.5 |
102 |
103 |
|
F5 |
86 |
24 |
2 |
15 |
3.5 |
102 |
104 |
|
F6 |
86 |
10 |
2 |
17 |
3.6 |
96 |
94 |
|
F7 |
72 |
10 |
2 |
18 |
3.3 |
94 |
97 |
|
F8 |
72 |
24 |
4 |
14 |
3.2 |
103 |
105 |
Study of Influence of Independent variables:
Disintegration Time (R1):
Polynomial equation in coded form was developed to assess how formulation factor affected Disintegration time.
Coded Equation: Disintegration Time (R1) = 16.50 - 2.00(B) - 1.00(BC)
Disintegration time reduces when increasing concentration of Cross povidone, according to negative coefficient of B. The interaction between Cross povidone & Sodium stearyl fumarate appears to aid in reducing disintegration time, as indicated by negative coefficient of BC. This aligns with contour and 3D surface plots [Figure:13], which visually depict the corelation between these variables and the Disintegration Time
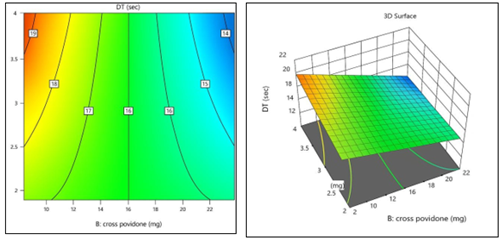
Figure 13:2D Contour plot & 3D Surface plot for Disintegration Time
Hardness (R2) :
Polynomial equation in coded form was developed to assess how formulation factor affected Hardness.
Hardness (R2) = 3.36 + 0.1375 (A) − 0.0125(B)
The positive coefficient of A (F-Melt® Type C) indicates that increasing its concentration leads to an increase in tablet hardness. The negligible and negative coefficient of B (Crospovidone) confirms its minimal effect. These results align with the 2D contour and 3D surface plots. [Figure14]
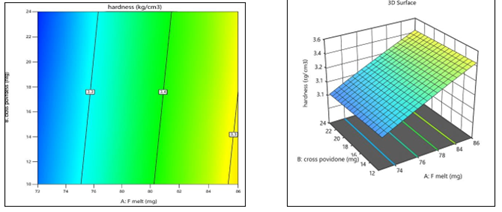
Figure 14: 2D Contour plot &3D Surface plot for Hardness
Drug release in Buffer (R3):
Polynomial equation in coded form was developed to assess how formulation factor affected %Drug Release in buffer
Drug release in Buffer (R3) = 99.62 + 2.88(B) + 0.8750(C) – 0.8750 (BC)
The positive coefficient of B (Cross Povidone) suggests that increasing its concentration enhances drug release, likely due to improved tablet porosity and disintegration. Similarly, C (Sodium stearyl fumarate) also shows a positive influence, though smaller in magnitude. The negative coefficient for the interaction term (BC) implies a slight antagonistic effect when both excipients are used together at higher levels, resulting in a mild reduction in drug release. These observations are confirmed by the 2D contour and 3D surface plots [Figure.15], which illustrate optimal drug release at moderate to high concentrations of Cross Povidone and Sodium Stearyl Fumarate used individually.
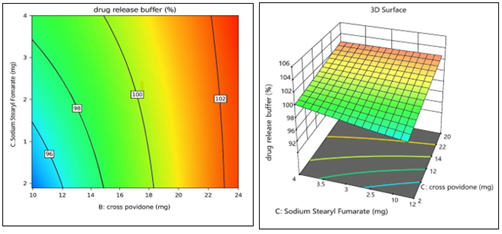
Figure15:2D Contour plot &3D Surface plot of %Drug Release in Buffer
Drug release in SSF(R4):
Polynomial equation in coded form was developed to assess how formulation factor affected %Drug Release in SSF
Drug release in SSF (R4) = 100.50 – 1.25(A) + 3.75 (B) + 0.5000 (C) – 0.7500(BC)
The positive coefficient of B (Cross Povidone) implies that increasing its concentration significantly enhances drug release, likely as a result of its rapid disintegration and wicking ability. The negative coefficient of A (F-Melt® Type C) suggests that higher concentrations of this excipient may slightly reduce drug release, possibly due to a stronger matrix formation delaying release. The positive coefficient of C (Sodium stearyl fumarate) indicates a mild enhancing effect, though statistically non-significant. The negative BC interaction term implies a slight antagonistic interaction when both Cross Povidone and Sodium stearyl fumarate are present at higher levels. These observations are confirmed by the 2D contour and 3D surface plots plots [Figure16].
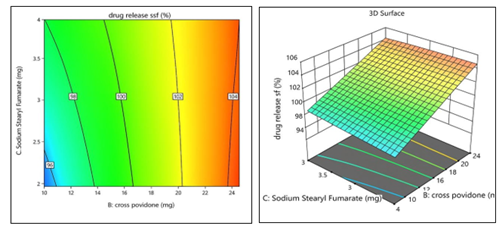
Figure 16: 2D Contour plot &3D Surface plot for %Drug Release in SSF
Evaluation of Tablet Properties of Optimized Batches:
Pre Compression Parameter
Table 6: Pre compression parameter results
|
Batches |
Bulk Density (gm/ml) |
Tapped Density (gm/ml) |
Carr's Index (%) |
Hausner's Ratio |
Angleof Repose (o) |
|
F1 |
0.44 ± 0.001 |
0.54±0.006 |
18.51±0.31 |
1.22 ±0.01 |
27±0.52 |
|
F2 |
0.69±0.004 |
0.75±0.003 |
8±0.34 |
1.1 ±0.08 |
27±0.58 |
|
F3 |
0.46±0.008 |
0.50±0.004 |
8±0.61 |
1.0 ±0.04 |
27±0.51 |
|
F4 |
0.47±0.012 |
0.54±0.005 |
12±0.45 |
1.14 ±0.09 |
23±0.47 |
|
F5 |
0.43±0.013 |
0.51±0.008 |
15±0.57 |
1.1±0.07 |
21±0.51 |
|
F6 |
0.49±0.007 |
0.54±0.009 |
9.25±0.44 |
1.1±0.03 |
28±0.52 |
|
F7 |
0.47±0.003 |
0.51±0.004 |
7.8±0.38 |
1.0±0.04 |
26±0.45 |
|
F8 |
0.44±0.005 |
0.47±0.003 |
6.38±0.41 |
1.06±0.02 |
25±0.49 |
Precompression parameters were applied to various powder ratios These results indicate that the formulation possesses suitable flow properties for further processing and tablet compression, which is critical for ensuring uniformity and consistency in the final dosage form.
Post-compression parameters:
Table 7: Post compression parameters results
|
Batches |
Weights (mg) |
Thickness (mm) |
Hardness (kg/cm2) |
Friability (%) |
Drug Content (%) |
|
F1 |
198±0.11 |
2.8±0.04 |
3.2±0.1 |
0.08±0.002 |
98±0.28 |
|
F2 |
200±0.38 |
3.0±0.03 |
3.4±0.1 |
0.2±0.003 |
98±0.39 |
|
F3 |
186±0.22 |
2.7±0.11 |
3.2±0.15 |
0.3±0.012 |
98.9±0.10 |
|
F4 |
214±0.54 |
3.2±0.04 |
3.5±0.1 |
0.08±0.033 |
98.8±1.00 |
|
F5 |
212±0.37 |
3.2±0.07 |
3.5±0.11 |
0.2±0.021 |
98.6±0.54 |
|
F6 |
198±0.21 |
2.8±0.01 |
3.6±0.18 |
0.20±0.001 |
98.5±2.14 |
|
F7 |
184±0.15 |
2.5±0.09 |
3.3±0.15 |
0.47±0.005 |
99.8±0.85 |
|
F8 |
200±0.38 |
3.0±0.08 |
3.2±0.1 |
0.10±0.007 |
99±1.00 |
The physicochemical tablet qualities of the optimization batches were assessed and are compiled in [Table 7], providing detailed information on parameters like weight, thickness, hardness, friability & drug content. Tablets ranged in 184 mg ± 0.15 to 214 mg ± 0.54 considered satisfactory. Tablets showed a mean hardness ranging from3.2 kg/cm² ± 0.1 and 3.6 kg/cm² ± 0.18, ensuring mechanical strength adequate during handling and packaging. The tablet’s thickness varied between 2.5 mm ± 0.09 to 3.2 mm ± 0.07, which was seen to be consistent and within satisfactory range for all batches. The tablet's Friability ranged0.08% ± 0.002 to 0.47% ± 0.005, with all batches remaining well below the 1% limit. Tablet formulations stayed within 1 % of the IP limit. All formulations had satisfactory Drug content uniformity, with values ranging from 98.0% ± 0.28 to 99.8% ± 0.85, indicating proper tablet component mixing. These results demonstrate that all evaluated batches exhibited acceptable physicochemical properties suitable for ODT formulations.
In-vitro Drug Release/Dissolution Studies:
All formulations studied showed immediate drug release over 30 min interval, promoting faster onset, therapeutic effects and enhancing patient compliance. This immediate release provides a more consistent drug level in the bloodstream improving overall treatment outcomes [Figure 17] demonstrated the release of drug (%) performed by Franz diffusion cell. [Figure 18,19] demonstrated release of drug (%) of 8 batches in Phosphate buffer pH 6.5 & simulated salivary fluid (SSF) pH 6.8.

Figure 17: In-vitro Dissolution with Franz diffusion cell
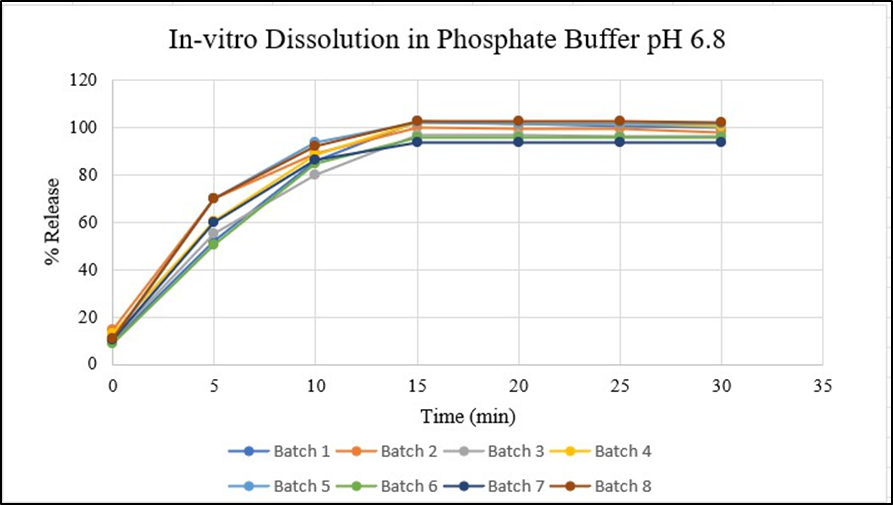
Figure18: In-vitro Dissolution of F1-F8 Batches in Phosphate Buffer pH6.8

Figure 19: In-vitro Dissolution of F1-F8 Batches in SSF
Interpretation of Overlay Plot
The design space is depicted using the plot[Figure 20], showing how input and process factors work together to guarantee quality. The area where Disintegration time, Hardness, % drug release in Buffer and % drug release in SSF are optimized is highlighted in the figure produced by the Design Expert Software. The optimal F- MELT TYPE C, Crospovidone, and sodium stearyl fumarate combination that produced the greatest results was discovered using plot analysis. It was found that 81.323mg of F-Melt® Type C, 11.152 mg of Crospovidone, and 3.419 mg of sodium stearyl fumarate were the combinations. A predicted Disintegration time (sec) of 17 sec, Hardness of 3.4 kg/cm2, % drug release in Phosphate buffer of 95.82 and % drug release in SSF of 95.97 were derived from this combination.
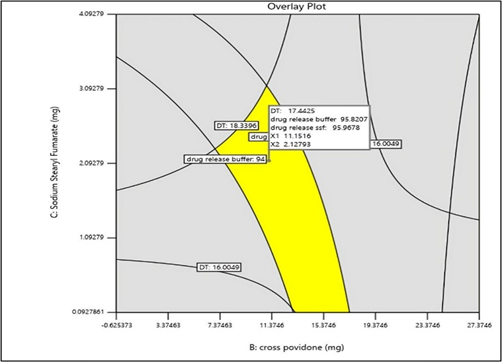
Figure 20: Overlay Plot of optimized Batch
Evaluation of the Optimized Formulation
Table 8: Results of Pre compression parameter of Optimised Batch
|
Sr.no |
Parameters |
Results |
|
1 |
Angle of Repose |
21°± 2.5 |
|
2 |
Bulk Density |
0.45 ± 0.04 gm/ml |
|
3 |
Tapped Density |
0.49± 0.02 g/ml |
|
4 |
Carr’s Index |
8.1± 2% |
|
5 |
Hausner’s Ratio |
1.0± 0.1 |
Table 9: Results of Post-compression parameters of Optimized Batch
|
Sr. no |
Parameters |
Results |
|
1 |
Thickness |
2.8± 0.2mm |
|
2 |
Hardness |
3.3 ± 0.1 kg/cm2 |
|
3 |
Friability |
0.05± 0.4 % |
|
4 |
Weight variation |
195.3 ± 1.5 mg |
|
5 |
Drug content |
99.67 ± 0.15%% |
|
6 |
Wetting Time |
20 ± 2 sec |
|
7 |
Disintegration time |
17± 2 sec |
|
8 |
Drug Release in Phosphate Buffer pH6.8 |
96 ± 2% |
|
9 |
Drug Release in SSF pH 6.8 |
97 ± 3% |
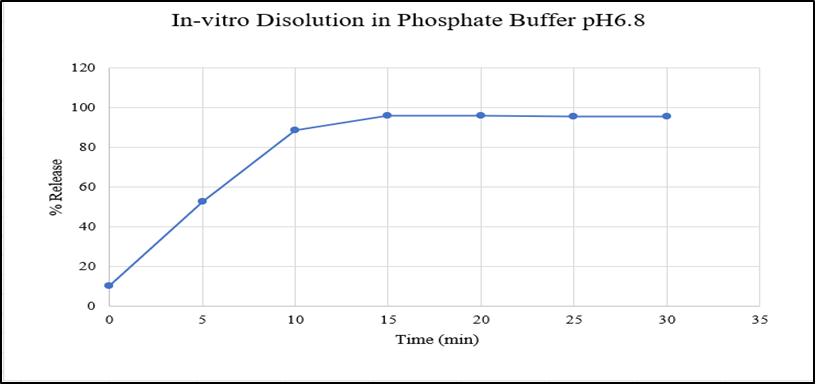
Figure 21: In-vitro Dissolution of optimized batch in Phosphate buffer pH6.8

Figure 22: In-vitro Dissolution of optimized batch in SSF pH6.8
Wetting Time Test

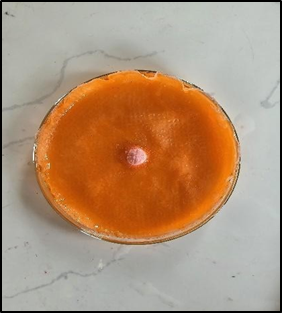
Figure 23: Wetting at start - 0 sec Figure 24: Wetting at end- 20 sec
Predicted & Actual Results of Selected Optimized Batch
Table 10: Predicted and Actual values for the optimized Levodopa Orally Disintegrating tablet
|
Batch no |
Factor 1 |
Factor 2 |
Factor 3 |
Disintegration time(sec) |
Hardness (kg/cm2) |
Drug Release in Buffer |
Drug Release in SSF |
|
Predicted |
81.323 |
11.152 |
2.128 |
17 |
3.4 |
95.82% |
95.97% |
|
Actual |
81.323 |
11.152 |
2.128 |
17 |
3.3 |
96% |
97% |
Short-term Stability Studies
After 1 month of stability studies, the optimized formulation tablets were checked for physical appearance, Hardness, Disintegration time, Drug release in Buffer & Drug release in SSFas shown in [Table 11]. During the trial, there was absence of noticeable change in the appearance of the Levodopa Orally Disintegrating tablets. The Disintegration time, Hardness, % Drug release in buffer & SSFpH6.8 remained consistent with the initial values, demonstrating that optimized formulation maintained its integrity. These results demonstrate the formulation's stability over the study period, ensuring that the therapeutic efficacy and release profile are preserved. The findings demonstrate that the formulation is reliable for long-term use and supports its potential for clinical application.
Table 11: Results of stability studies
|
Sr. no |
Parameters |
Observations |
||
|
Before Stability |
After Stability of 1 month |
|||
|
|
|
|
At accelerated temp 40°C ± 2°C |
At Room Temp 37± 2°C |
|
1 |
Appearance |
Off white |
Off white |
Off white |
|
2 |
Disintegration time |
17sec |
17sec |
17sec |
|
3 |
Hardness |
3.3kg/cm2 |
3.3 kg/cm2 |
3.3 kg/cm2 |
|
4 |
Drug release in Buffer |
96% |
95% |
96% |
|
5 |
Drug release in SSF |
97% |
96% |
97% |
CONCLUSION
A patient-focused Orally Disintegrating Tablet (ODT) of Levodopa was successfully developed using F-Melt® Type C. This formulation aims to quick relief from symptoms of Parkinson’s disease, also help people who experience difficulty in swallowing. A 23 full factorial design was utilized to optimize important formulation factors. The result was tablets that disintegrate quickly (about 17 seconds), release a high percentage of levodopa drug (around 97% in simulated salivary fluid and ~96% in phosphate buffer pH 6.8), have enough mechanical strength, and taste good. Stability studies demonstrated that the chemical and physical propertiesmet ICH guidelines. Unlike other Levodopa ODTs, this formulation does not contain phenylalanine, making it safer for patients with PKU. Overall, this optimized Levodopa ODT has strong potential to enhance patient adherence and therapeutic outcomes, the successful development of this Levodopa ODT formulation using F-Melt® Type C as a co-processed excipient on large scale will be an excellent opportunity in the treatment of symptoms of Parkinson Disease.
ACKNOWLEDGEMENTS
I would like to express my heartfelt gratitude to first and foremost my research guide, Mrs. Rachana Sarawade, for invaluable guidance and support throughout the project. I am grateful to Molychem Pvt Ltd, Mumbai for providing drug i.e. Levodopa, Mrs. Avinash Phadatare, Transchem Coorporation Pharma Pvt Ltd, for providing Ludiflash, Fuji Chemical/Barentz India for providing F-Melt® Type C, Umedica Laboratories Pvt.Ltd. for providing Sodium stearyl fumarate and My sincere thanks to to Shri.C.B.Patel Research Centre for Chemistry and Biological Sciences Mumbai for analyzing my DSC sample. B.K Birla College of Arts,Science and Commerce (Autonomous), Kalyan Nanotech Research Laboratory for analyzing my XRD samples,I want to express my gratitude to our Principal, Dr.Paraag Gide, Dr. L H Hiranandani College of Pharmacy, Ulhasnagar-03 for his generous consideration and facilities and of course for his constant support that made this research possible. I would like to express my sincere gratitude to all those who contributed to the success of this research article Thank you all!
Relevant conflicts of interest/financial disclosures: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
REFERENCES


Shraddha Raorane, Rachana Sarawade, Formulation, Development and Evaluation of Orally Disintegrating Tablet of Levodopa for Parkinson’s Disease, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 1, 1649-1669. https://doi.org/10.5281/zenodo.1827289
 10.5281/zenodo.1827289
10.5281/zenodo.1827289
