Department of Pharmaceutical Chemistry, Mar Dioscorus College of Pharmacy, Alathara, Thiruvananthapuram.
Inflammatory diseases are widespread and affect millions of people worldwide. Current treatments of inflammatory diseases have limited efficacy and significant side effects. So an attempt is to be made to explore the anti-inflammatory activity of 1,3,4-oxadiazole. A novel series of 1,3,4-oxadiazole derivatives bearing imidazole moiety were designed using ACD Lab/ Chemsketch 12.0 and their properties were predicted using the Molinspiration software and they obeyed the Lipinski rule of five. The designed derivatives showing optimal physicochemical properties were selected for docking studies Docking studies of these compounds were performed on cox-2 (5IKQ) using Auto Dock Vina. The result obtained from the docking study revealed that compounds a35, a36, a26 showed good activities on cox-2. Overall, the study concluded that 1,3,4-oxadiazole derivatives bearing imidazole moiety could be considered as promising for the development of novel anti-inflammatory agents.
Drug discovery aims at identifying a compound therapeutically useful in treating and curing a disease. Normally the drug discovery process starts with a biological target that has been shown to play a role in the development of the disease or begins from a molecule with interesting biological activities. The drug discovery process involves the identification of candidates, synthesis, characterization, screening, and assays for therapeutic efficacy. Once a compound has shown its value in these tests, it will begin the process of drug development prior to clinical trials. Drug discovery involves the combination of many disciplines and interests starting from a simple process of identifying an active compound.
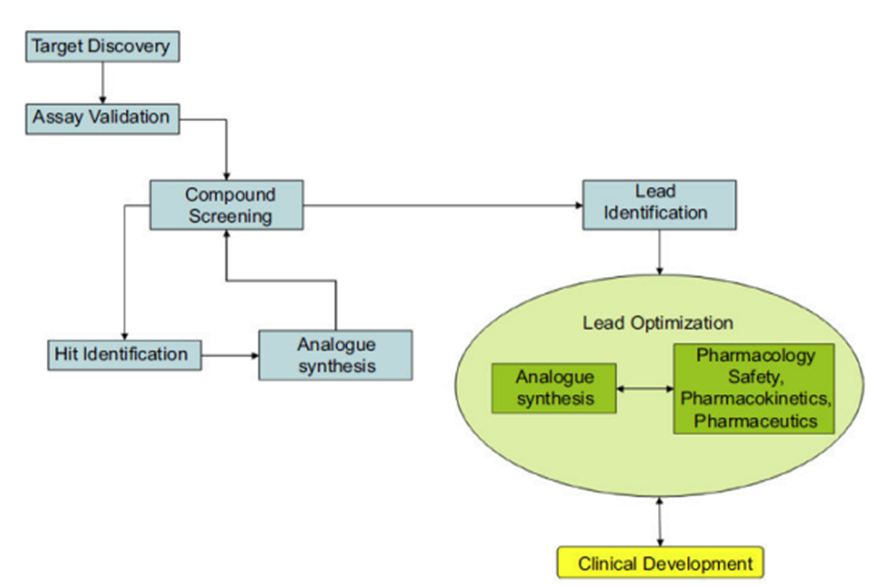
Fig.1. General process of drug discovery
The first step in the drug development process is the identification of a new chemical entity that modifies a cell or tissue function. Once it shown to be effective and selective, a compound which is to be discovered must be completely free of toxicity, should have good bioavailability and marketable before it can be considered to be a therapeutic entity. Drug design is a creative science, a special technology, and an art all in one. [1][2][3][4][5] Azoles are nitrogen, sulfur and oxygen containing compounds with a five-membered ring system that comprises thiadiazole, oxadiazole, triazole, imidazole, pyrazole and other rings. These compounds exhibit wide range of medicinal applications in the treatment of various types of diseases. Imidazole have occupied a unique position in heterocyclic chemistry, and its derivatives have attracted considerable interests in recent years for their versatile properties in chemistry and pharmacology. Imidazole is nitrogen-containing heterocyclic ring which possesses biological and pharmaceutical importance. Thus, imidazole compounds have been an interesting source for researchers for more than a century.[6] Oxadiazole have four possible isomer forms but 1,3,4-oxadiazole is widely explored for various applications. 1,3,4-oxadiazoles are the heterocyclic compounds containing one oxygen and two nitrogen atom in a five membered ring which posses wide range of biological activities such as antibacterial, antifungal, anti-inflammatory and anticancer activity. The ring in 1,3,4-oxadiazole is aromatic due the presence of conjugated pi electrons. This gives it stability and influences its reactivity.[7][8]
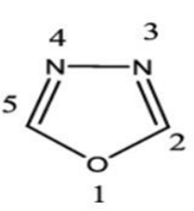
Fig.2. 2D Structure Of 1,3,4-Oxadiazole
When cells are damaged, the body's defense mechanism, inflammation, causes immune cells to proliferate in an attempt to eradicate the stimulus. Prolonged inflammation, however, is harmful and can result in a variety of pathological conditions, including cancer, atherosclerosis, arthritis, and neurological diseases. Phospholipase A2 activation initiates the inflammatory response by causing the lipid bilayer of the cell membrane to release arachidonic acid (AA). Cyclooxygenase enzymes (COX-1 and COX-2) quickly break down AA into prostaglandin H2 (PGH2), which is then transformed into eicosanoids like prostaglandin (PG), prostacyclin (PGI2), and thromboxanes (TXA2). These eicosanoids control the production of inflammatory cytokines and the emergence of common inflammation symptoms like fever, redness, swelling, and pain. 1,3,4- oxadiazole have been found to exhibit potential anti-inflammatory activity. So novel drugs needs to be developed to overcome the treatment failures.[9][10][11][12][13] In this study, we have designed and evaluated a series of new 1,3,4-Oxadiazole derivatives of 3-[(substituted)methyl]-5- [(1H- imidazole-1-yl)4-methyl] -1,3,4-oxadiazole -2(3H)- thione ,in search of potent anti-inflammatory agents through In-silico studies.
Scheme Of Work [14]
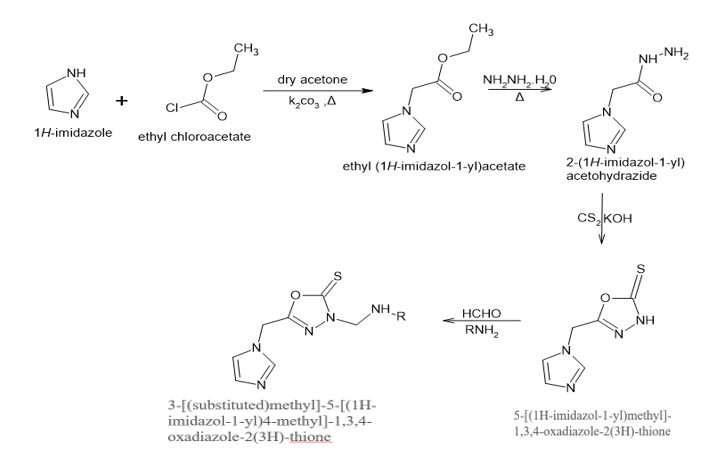
MATERIALS AND METHODS
ACD Lab Chemsketch
ACD/ChemSketch is a molecular modeling application that allows users to create and edit chemical structure images. It also has capabilities like molecular property calculations (e.g., molecular weight, density, molar refractivity, etc.), cleaning and viewing of 2D and 3D structures, naming structures (less than 50 atoms and 3 rings), and logP prediction. Using ACD Labs Chemsketch version 12.0, the compounds' chemical structures and SMILES notations were acquired.[16]
ACD/ChemSketch has the following major capabilities:
· Structure Mode for drawing chemical structures and calculating their properties.
· Draw Mode for text and graphics processing.
· Molecular Properties calculations for automatic estimation of formula weight, percentage composition, molar refractivity, molar volume, parachor, surface tension, density, dielectric constant, polarizability
Molinspiration
Molinspiration is an independent research organization focused on development and application of modern cheminformatics techniques, especially in connection with the internet. It offers broad range of cheminformatics software tools supporting molecule manipulation and processing, including SMILES and SD file conversion, normalization of molecules, generation of tautomers, molecule fragmentation, calculation of various molecular properties needed in QSAR, molecular modelling and drug design, high quality molecule depiction, molecular database tools supporting substructure search or similarity and pharmacophore similarity search. SMILES notations of the selected derivatives were fed in the online Molinspiration software (https://www.molinspiration.com/) to predict the drug likeness properties. Lipinski’s rule of five is used in drug design and development to predict oral bioavailability of potential lead or drug molecules. Lipinski rule is also known as Pfizers rule of five / Lipinski’s rule of 5. The rule was formulated by the scientist Christopher A Lipinski. The Lipinski rule of five states that an orally active drug should obey the following criteria: 1. Not more than five hydrogen bond donors 2. Not more than 10 hydrogen bond acceptors 3. Molecular weight less than 500 Daltons 4. An octanol-water partition coefficient log P not greater than 5 5. Not more than 5 rotatable bonds.[17]
PASS (Prediction of Activity Spectra for Substances)
PASS is a program used for calculating sample sizes or determining the statistical power of a test or confidence interval in research studies. NCSS LLC is the company that produces PASS. It is a comprehensive software package with over 290 documented sample size and power procedures. It’s widely recognized as the leading tool for determining sample sizes in various fields such as clinical trials, pharmaceuticals, and medical research. Inputting the structural formula of a drug-like substance into specialized software can provide an estimated biological activity profile as an output.
Protein Data Bank
The Protein Data Bank (PDB) is a online database that stores three-dimensional structural data of large biological molecules, including proteins and nucleic acid. This data is gathered using techniques like X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy. Scientists worldwide contribute to the database, and the information is freely accessible through member organization websites like PDBe, PDBj, RCSB and BMRB. The Protein Data Bank (PDB)is overseen by the Worldwide Protein Bank(wwPDB), an organization responsible for managing and maintaining the database. The PDB plays a crucial role in structural biology, particularly in areas like structural genomics. Many top scientific journals and funding agencies now mandate scientists to submit their structural data to the Protein Data Bank (PDB).[18][19][20]
Protein preparation
The typical structure file from the PDB may require preprocessing before it can be readily utilized in molecular modelling calculations. It consists of only heavy atoms and may include a co-crystallized ligand, water molecules, metal ions, activators, cofactors and several protein subunits. According to the requirement protein is pre-processed, optimized and minimized with forcefield. Optimization involves water removal and minimization. The protein structure was refined. The ionization structure and most stable were chosen.
Ligand preparation
Ligand preparation involved energy of the ligands. Ligprep [Schriinger, 2010] facilitated this process by adding hydrogen, converting 2D structures to 3D, generating stereoisomers, and optionally neutralizing charged structures or determining the most probable ionization state at a user-defined PH. All the structures were ionized at a neutral pH of 7. Conformers for each ligand were generated using Conf Gen, applying the OPLS-2005 force field method.
Docking
Molecular docking is a structure-based computational method that generates the binding mode and affinity between ligands and targets by predicting their interactions [18, 19]. There are several docking tools used for this purpose. AutoDock, AutoDock Vina, GOLD, Glide, MOE, ICM, and FlexX are amongst the popular software in use. Molecular docking has various applications in the drug discovery and design process. In the early years of its application, it was mainly used to investigate the molecular interactions between ligands and targets. [21][22]
AutoDock
AutoDock comprises 3 C programs, AutoTors, responsible for refining ligand input, AutoGrid, which calculates interaction energies using macromolecular coordinates, and AutoDock itself, tasked with the actual docking process. AutoDock effectively reproduces ligand positions by considering six spatial degrees of freedom-rotation and translation. It accurately captures crystallographically determined positions for ligands with up to eight torsional degrees of freedom. It’s simulated annealing search may struggle to adequately explore conformational space for molecules with a high number of degrees of freedom, potentially limiting its applicability in certain cases. [23][24]
RESULT AND DISCUSSION
Fifty analogues of 1,3,4-Oxadiazole derivatives were designed using ACD Lab Chemsketch 12.0. Initially the designed fifty analogues were subjected to Lipinski rule analysis using molinspiration software.
Theoretical determination of drug-likeness properties
We predicted the drug likeliness profile of the compounds through analysis of pharmacokinetic properties of the compounds by using molinspiration online software. Based on the results obtained from molinspiration it was observed that all of the proposed compounds obeyed Lipinski rule of five. According to the Lipinski’s rule of five new molecule designed for oral route should have a MW < 500, log P o/w < 5, No more than 5 hydrogen bond donors and No more than 10 hydrogen bond acceptor. From the Lipinski rule analysis, all the 50 compounds were selected for further studies, since the compound did not show any violations from the Lipinski rule of five. Structure of proposed 1,3,4- oxadiazole derivatives of 3-[(substituted)methyl]-5- [(1H- imidazole-1-yl)4-methyl] -1,3,4-oxadiazole -2(3H)- thione is shown in Figure 3.
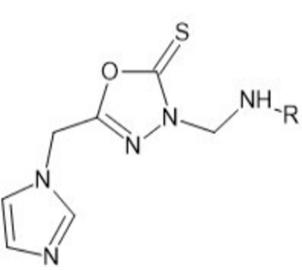
Figure 3: Designed Ligand
The results of Lipinski rule analysis of first 50 compounds are shown in the table 1.
Table 1: Lipinski Rule Analysis of Proposed Derivatives
|
Compound Code |
R |
Mw <500 |
Logp <5 |
Nhdon<5 |
Nhacc<10 |
Nrotb<10 |
N Violation |
|
a1 |
C6H5 |
293.40 |
1.11 |
3 |
6 |
5 |
0 |
|
a2 |
C6H5OCH3 |
323.42 |
1.15 |
3 |
7 |
6 |
0 |
|
a3 |
C6H5OC2H5 |
3337.45 |
1.52 |
3 |
7 |
7 |
0 |
|
a4 |
C6H5F |
314.30 |
1.22 |
1 |
6 |
5 |
0 |
|
a5 |
C6H5F |
305.34 |
1.27 |
1 |
6 |
5 |
0 |
|
a6 |
C6H5F |
305.34 |
1.25 |
1 |
6 |
5 |
0 |
|
a7 |
C6H5CH3 |
301.38 |
1.55 |
1 |
6 |
5 |
0 |
|
a8 |
C6H5Cl |
321.79 |
1.74 |
1 |
6 |
5 |
0 |
|
a9 |
C6H5Cl |
321.79 |
1.78 |
1 |
6 |
5 |
0 |
|
a10 |
C6H5Br |
366.24 |
1.87 |
1 |
6 |
5 |
0 |
|
a11 |
C6H5Br |
366.24 |
1.92 |
1 |
6 |
5 |
0 |
|
a12 |
C6H5Br |
366.24 |
1.89 |
1 |
6 |
5 |
0 |
|
a13 |
C6H5Cl |
321.79 |
1.76 |
1 |
6 |
5 |
0 |
|
a14 |
C6H5CH3 |
301.38 |
1.51 |
1 |
6 |
5 |
0 |
|
a15 |
C6H5CH3 |
301.38 |
1.53 |
1 |
6 |
5 |
o |
|
a16 |
C6H5OCH3 |
317.37 |
1.11 |
1 |
7 |
6 |
0 |
|
a17 |
C6H5OCH3 |
317.37 |
1.16 |
1 |
7 |
6 |
0 |
|
a18 |
C6H5OCH3 |
317.37 |
1.14 |
1 |
7 |
6 |
0 |
|
a19 |
C6H5OC2H5 |
331.40 |
1.49 |
1 |
7 |
7 |
0 |
|
a20 |
C6H5OC2H5 |
331.40 |
1.54 |
1 |
7 |
7 |
0 |
|
a21 |
C6H5OC2H5 |
331.40 |
1.51 |
1 |
7 |
7 |
0 |
|
a22 |
C6H5NH2 |
302.36 |
0.54 |
3 |
7 |
5 |
0 |
|
a23 |
C6H5NH2 |
302.36 |
0.18 |
3 |
7 |
5 |
0 |
|
a24 |
C6H5NH2 |
302.36 |
0.16 |
3 |
7 |
5 |
0 |
|
a25 |
C6H5NO2 |
332.35 |
1.02 |
1 |
9 |
6 |
0 |
|
a26 |
C6H5NO2 |
332.35 |
1.04 |
1 |
9 |
6 |
0 |
|
a27 |
C6H5NO2 |
332.35 |
1.06 |
1 |
9 |
6 |
0 |
|
a28 |
C6H5OH |
303.35 |
0.84 |
2 |
7 |
5 |
0 |
|
a29 |
C6H5OH |
303.35 |
0.60 |
2 |
7 |
5 |
0 |
|
a30 |
C6H5OH |
303.35 |
0.63 |
2 |
7 |
5 |
0 |
|
a31 |
C6H5(OH)2 |
319.35 |
0.14 |
3 |
8 |
5 |
0 |
|
a32 |
C6H5(OH)2 |
319.35 |
0.34 |
3 |
8 |
5 |
0 |
|
a33 |
C6H5(NH2)2 |
317.38 |
-0.14 |
5 |
8 |
5 |
0 |
|
a35 |
C6H5(Cl)3 |
390.68 |
3.00 |
1 |
6 |
5 |
0 |
|
a36 |
C6H5(Cl)3 |
390.68 |
3.00 |
1 |
6 |
5 |
0 |
|
a37 |
C6H5(Cl)2 |
356.24 |
2.39 |
1 |
6 |
5 |
0 |
|
a38 |
C6H5(Cl)2 |
356.24 |
2.39 |
1 |
6 |
5 |
0 |
|
a39 |
C6H5(Cl)2 |
356.24 |
2.39 |
1 |
6 |
5 |
0 |
|
a40 |
C6H5(F)2 |
323.33 |
1.36 |
1 |
6 |
5 |
0 |
|
a41 |
C6H5(F)2 |
323.33 |
1.36 |
1 |
6 |
5 |
0 |
|
a42 |
C6H5(F)2 |
323.33 |
1.36 |
1 |
6 |
5 |
0 |
|
a43 |
C6H5(Br)2 |
445.14 |
2.65 |
1 |
6 |
5 |
0 |
|
a44 |
C6H5(Br)2 |
445.14 |
2.65 |
1 |
6 |
5 |
0 |
|
a45 |
C6H5(Br)2 |
445.14 |
2.65 |
1 |
6 |
5 |
0 |
|
a46 |
C6H5(Br)2 |
445.14 |
2.65 |
1 |
6 |
5 |
0 |
|
a47 |
C6H5(F)2 |
323.33 |
1.36 |
1 |
6 |
5 |
0 |
|
a48 |
C6H5(Cl)2 |
356.24 |
2.39 |
1 |
6 |
5 |
0 |
|
a49 |
C6H5(OH)2 |
319.35 |
0.34 |
3 |
8 |
5 |
0 |
|
a50 |
C6H5(CH3)2 |
315.40 |
1.93 |
1 |
6 |
5 |
0 |
PASS (Prediction of Activity Spectra for Substances)
The PASS software predicts the biological activity of proposed derivatives where Pa indicates probability to be active and Pi indicates probability to be inactive. The PASS value of proposed derivatives were shown in table 2.
Table 2: PASS of designed 50 compounds
|
Compound code |
Pa |
Pi |
|
a1 |
0,372 |
0,128 |
|
a2 |
0361 |
0140 |
|
a3 |
0365 |
0135 |
|
a4 |
0288 |
0223 |
|
a5 |
0289 |
0222 |
|
a6 |
0135 |
0087 |
|
a7 |
0344 |
0158 |
|
a8 |
0288 |
0224 |
|
a9 |
0313 |
0193 |
|
a10 |
0124 |
0035 |
|
a11 |
0063 |
0028 |
|
a12 |
0056 |
0036 |
|
a13 |
0282 |
0232 |
|
a14 |
0343 |
0159 |
|
a15 |
0313 |
0193 |
|
a16 |
0350 |
0151 |
|
a17 |
0390 |
0111 |
|
a18 |
0361 |
0140 |
|
a19 |
0354 |
0147 |
|
a20 |
0394 |
0107 |
|
a21 |
0365 |
0135 |
|
a22 |
0060 |
0031 |
|
a23 |
0063 |
0028 |
|
a24 |
0056 |
0036 |
|
a25 |
0351 |
0150 |
|
a26 |
0362 |
0138 |
|
a27 |
0391 |
0110 |
|
a28 |
0394 |
0107 |
|
a29 |
0409 |
0093 |
|
a30 |
0435 |
0072 |
|
a31 |
0367 |
0134 |
|
a32 |
0382 |
0118 |
|
a33 |
0054 |
0038 |
|
a34 |
0059 |
0033 |
|
a35 |
0334 |
0164 |
|
a36 |
0381 |
0120 |
|
a37 |
0056 |
0036 |
|
a38 |
0061 |
0030 |
|
a39 |
0282 |
0232 |
|
a40 |
0053 |
0039 |
|
a41 |
0058 |
0034 |
|
a42 |
0058 |
0033 |
|
a43 |
0054 |
0038 |
|
a44 |
0061 |
0030 |
|
a45 |
0125 |
0034 |
|
a46 |
0055 |
0036 |
|
a47 |
0271 |
0246 |
|
a48 |
0400 |
0102 |
|
a49 |
0343 |
0159 |
|
a50 |
0,157 |
0,072 |
|
Standard (Phenylbutazone) |
0961 |
0030 |
Docking
From the docking result of 50 derivatives 25 derivatives which shows high and similar docking score than standard drug is shown in the table.
Table 3: Docking score
|
Compound? Code? |
R Group |
Docking? Score? |
|
a5? |
C6H5F |
-7.1? |
|
a7? |
C6H5CH3 |
-7.2? |
|
a8? |
C6H5Cl |
-7.0? |
|
a9? |
C6H5Cl |
-7.0? |
|
a12 |
C6H5Br |
-7.1? |
|
a13 |
C6H5Cl |
-7.0? |
|
a14 |
C6H5CH3 |
-7.1? |
|
a15 |
C6H5CH3 |
-7.1? |
|
a25 |
C6H5NO2 |
-7.2? |
|
a26 |
C6H5NO2 |
-7.5? |
|
a27 |
C6H5NO2 |
-7.2? |
|
a35 |
C6H5(Cl)3 |
-7.7? |
|
a36 |
C6H5(Cl)3 |
-7.5? |
|
a37 |
C6H5OH |
-7.1? |
|
a38 |
C6H5(Cl)2 |
-7.1? |
|
a39 |
C6H5(Cl)2 |
-7.0? |
|
a40 |
C6H5(F)2 |
-7.0? |
|
a41 |
C6H5(F)2 |
-7.0? |
|
a45 |
C6H5(Br)2 |
-7.0? |
|
a49 |
C6H5(OH)2 |
-7.3? |
|
a3? |
C6H5OC2H5 |
-6.9? |
|
a20? |
C6H5OC2H5 |
-6.9? |
|
a1? |
C6H5 |
-6.7? |
|
a31 |
C6H5(OH)2 |
-6.9? |
|
Standard (Phenylbutazone)? |
-7.1? |
|
Target ID :5IKQ
Protein: cox-2
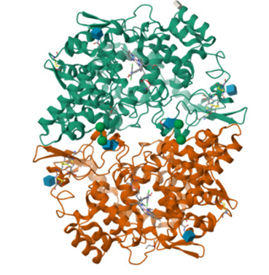
Figure 7: 3D Struture of cox-2
ompound a1
Among the 25 derivatives docked images of 13 derivatives were shown below.
Table 4: Docked images of 13 compounds showing high score
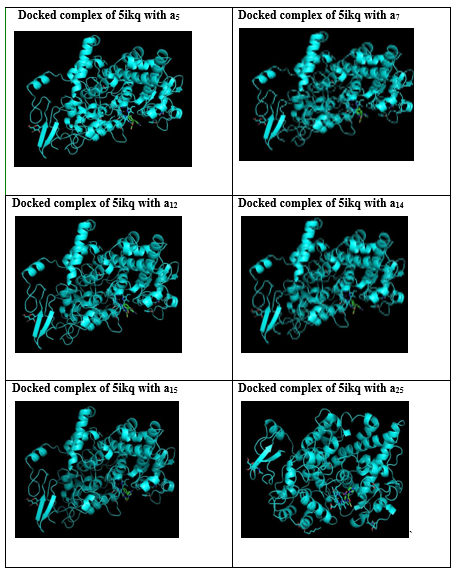
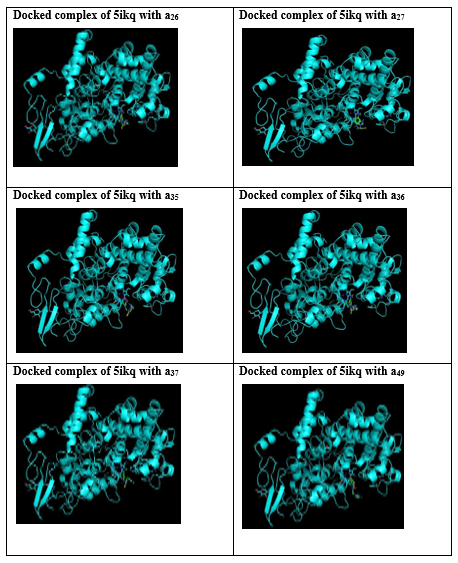
SUMMARY AND CONCLUSION
Summary
The present work deals with the synthesis of certain novel 1,3,4-oxadiazole derivatives and exploring their anti-inflammatory activities. The investigation that has been carried out is summarized below under separate headings.
i. Insilico designing of novel molecules using ACD Lab Chemsketch 12.0
ACD Lab Chemsketch 12.0 freeware software was used to design 50 1,3,4-oxadiazole derivatives.
ii. Molinspiration
50 novel 1,3,4-oxadiazole derivatives were designed by ACD/ Lab Chemsketch were subjected to calculation of “Lipinski’s rule of five” and drug likeness using molinspiration software.
iii. Docking studies
The compounds obtained above were subjected to docking using AutoDock Vina against the selected target protein 5IKQ (anti-inflammatory activity).Based on the docking score,compounds a1-a50 having docking score ranging from -6.1 to -7.7 for docking against cox-2 enzyme were identified.From this synthetically feasible compound a1 was selected for subsequent synthesis.
CONCLUSION
A novel series of 1,3,4-oxadiazole derivatives bearing imidazole moiety were designed using ACD Lab/ ChemSketch 12.0 and their properties were predicted using the Molinspiration software and they obeyed the Lipinski rule of five. The designed derivatives showing optimal physicochemical properties were selected for docking studies. Docking studies of these compounds were performed on cox-2 (5IKQ) using AutoDock Vina. The result obtained from the docking study revealed that compounds a35 (2,4,5-trichloroanilino methyl derivative of 1,3,4-oxadiazole), a36(2,4,6-trichloroanilino methyl derivative of 1,3,4-oxadiazole), a26(3-niroanilino methyl derivative of 1,3,4-oxadiazole) showed good activities on cox-2. Result of In-silico studies conclude that the majority of the compounds exhibited excellent drug likeness properties and favorable pharmacokinetic profiles. From the above findings, 1,3,4-oxadiazole derivatives bearing imidazole moiety could be identified as lead moieties for future work in the areas of anti-inflammatory development.
ACKNOWLDGMENT
The authors are thankful to the MAR DIOSCROUS COLLEGE OF PHARMACY management, Thiruvananthapuram, for providing all facilities for the present investigation.
REFRENCES






Rachel Mathew*, V. S. Anjana, S. Nakshathra, R. S. Shahana, A. P. Sona Nair, A. Vaheeda, Computational Design and Anti-Inflammatory Assessment of Novel 1,3,4-Oxadiazole Derivatives, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 3, 2055-2067. https://doi.org/10.5281/zenodo.15063808
 10.5281/zenodo.15063808
10.5281/zenodo.15063808
