Department of Pharmacology, Progressive Education Societys Modern College of Pharmacy, Yamunanagar Nigdi-411044, Pune, Maharashtra, India.
Biosimilars, which are biologic drugs that mimic reference biologics that have already received approval, are transforming contemporary medicine. Biologics and biosimilars are made from living organisms, which makes their synthesis more difficult and results in complex structures, in contrast to traditional small-molecule medications. Government incentives, the expiration of the major biologics' patents, and the possibility of large healthcare cost savings are some of the reasons behind the rapid growth of the global biosimilar business. The market for biosimilars is anticipated to reach $66.9 billion by 2028, with oncology, immunology, and endocrinology as the main therapeutic areas. Biosimilars promote competition and improve patient access by providing affordable substitutes for pricey reference biologics. The FDA and EMA are in charge of regulatory frameworks that offer strong yet adaptable rules to guarantee safety, effectiveness, and traceability. By 2024, biosimilars may save the US healthcare system $250 billion. The development process can cost up to $300 million over nine years, making it hard for many businesses to enter. Biosimilars have already made healthcare more affordable and available. extended. Biosimilars have already increased access to and affordability of healthcare. The long-term viability and widespread adoption of biologic drugs depend on consistent regulation, international collaboration, and ongoing innovation to fulfill the rising need for affordable biologics.
Biological agents differ from conventional medications in that they are derived from living systems and have larger molecules, more complicated structures, and species specificity. Biological agent therapy has revolutionized the treatment of various illnesses, including hormone replacement therapy, inflammatory conditions, and cancer [6], [24]. Approximately 10% of pharmaceutical spending is allocated to biological agents, a rapidly expanding segment of the pharmaceutical market that is projected to grow at an exponential rate. One pharmaceutical corporation began producing biological agents similar to generic medications when their patents were about to expire. Because of several factors, such as the utilization of a biological process system, cell lines, and their inherent natural variability, the manufacture and analysis of biological agents are more complex and technically harder than those of typical small-molecule agents made by chemical processes [24], [22]. These substances are known as "biosimilars," which are biological medications that contain a form of the active ingredient of a biologic that has previously received approval from its original manufacturer [5]. Similar biological medical goods, follow-on biologics, following-entry biologics, and similar biotherapeutic products are alternate names for biosimilars.
The biosimilar market
In 2006, the European Union authorized the first biosimilar for the growth hormone somatropin [2]. Also, since the request for biosimilars has grown, so-called alternate generation biosimilars, which include emulsion proteins and monoclonal antibodies, have surfaced [6]. The EU has the most sanctioned biosimilars and extensive knowledge of their efficacy and safety. A fairly new class of pharmaceutical products, biosimilars are anticipated to account for 210 billion (17.5% of all medical expenditures). As patents expire, the request for biologics in cancer across all suggestions is prognosticated to reach $68 billion by 2020. 8. Global deals of biosimilars reached over $150 billion in 2013 and $228 billion in 2016 and are anticipated to reach $390 billion by 2020 [10], [25]. Pharmaceutical companies looking for strategic benefits have turned their attention to the biosimilar geography, which sits at the nexus of profitable considerations, scientific complications, and nonsupervisory nuances. One of the biotechnology industry's swiftly growing subsectors is biopharmaceuticals. In particular, the biosimilar request is growing rapidly, with more than 200 authorized biosimilars worldwide as of right now. With a composite periodic growth rate (CAGR) of 17.8, the global biosimilars market is prognosticated to rise from its estimated $29.4 billion in 2023 to $66.9 billion by 2028. Grounded on tab prices, the US biologics industry has grown at an average annual pace of 12.5 over the last five years. Biologics now make up 46% of total spending, and this rise is faster than that of non-biologics [1]. Atex-manufacturer tab costs: The United States spent $568 billion for specifics in 2021. Biologics entered $260 billion of this sum, or 46% of the total quantum spent on specifics. A steady increase in the blessing of biosimilars has been observed following the patent expiration of certain inventor medicinals and the growing support for faster blessing paths from crucial non-supervisory bodies such as the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA). These accelerated paths are really a strong incitement for pharmaceutical companies looking to produce biosimilars, though indeed they aren't yet completely optimized for resource optimization [2]. The implicit offer of affordable backups for precious reference biologics is the primary profitable driver behind the development of biosimilars. Cases can go to the biosimilars since they've lower development costs than the reference drugs, which is particularly important for chronic illnesses. Likewise, biosimilars strategically position medicinal products to increase their market share, especially after inventor biologics' patents expire. By 2023, some 71 birth patents are anticipated to expire, offering challengers who want to produce and vend biosimilars a $55 billion profit eventuality.
Companies can strategically embrace biosimilar development to gain a footing in a variety of markets, meeting a range of healthcare requirements and promoting global health equity. Biosimilars are seen as powerful weapons for worldwide market penetration. This strategy improves competitiveness in a sector marked by quick scientific breakthroughs and changing market dynamics in addition to strengthening the company's market presence. As more and more products become accessible as a result of increasing rivalry, manufacturers are being forced to lower their prices in order to preserve or grow their market share. Additionally, pharmaceutical companies frequently choose strategic alliances and collaborations because they understand the logistical and technical challenges involved in the creation of biosimilars. By utilizing specialist knowledge, pooling resources, and working together to negotiate the complexities of biosimilar development, these synergies enable businesses to reduce risks and maximize efficiency. Biosimilars now have a huge chance to join the market thanks to this [21].
|
Parameter |
Data / Value |
|
Market size (2023) |
USD 29.4 billion |
|
Projected market size (2028) |
USD 66.9 billion |
|
CAGR (2023–2028) |
17.8% |
|
Number of approved biosimilars worldwide |
>200 |
|
Biologics share in total drug spending (2021, USA) |
46% of total USD 568 billion |
|
Fundamental growth drivers |
Patent expiry, regulatory support, cost savings |
Products that have been authorized by the US FDA as biosimilars and those that are being developed by various businesses at different stages
The biosimilar market is still growing, with both approved medications and attractive candidates in development for a number of biologic drugs. The authorized biosimilars for Filgrastim (Amgen's reference product NEUPOGEN) include RELEUKO (Amneal), ZARXIO (Sandoz), and NIVESTYM (Pfizer). Products from the pipeline include LUPIFIL (Lupin, Phase 1), TX01 (Tanvex, pending), and GRASTOFIL (Accord-Apotex, pending). APO-EPO (Apotex) is presently undergoing Phase 3 studies, while RETACRIT (Pfizer-Vifor) is an approved biosimilar of epoetin (marketed as EPOGEN by Amgen and PROCRIT by Johnson & Johnson). A number of biosimilars, such as FULPHILA (Mylan), UDENYCA (Coherus), ZIEXTENZO (Sandoz), NYVEPRIA (Pfizer), STIMUFEND (Fresenius), and FYLNETRA (Amneal), have already received approval for pegfilgrastim (NEULASTA by Amgen). The development pipeline includes TX04 (Tanvex, Phase 1), LUPIFIL-P (Lupin, pending), and LAPELGA (Accord-Apotex, pending). There are no known pipeline candidates for insulin glargine (LANTUS by Sanofi), but there are two licensed biosimilars: SEMGLEE (Viatris-Mylan) and REZVOGLAR (Eli Lilly). While XLUCANE (Stada, Phase 3) and LUBT010 (Lupin, Phase 3) are being developed, biosimilars such as BYOOVIZ (Biogen) and CIMERLI (Coherus) have been approved for ranibizumab (LUCENTIS by Genentech). Lastly, there is a strong pipeline for Aflibercept (EYLEA by Regeneron), but there aren't any approved biosimilars at this time. ABP 938 (Amgen, Phase 3), FYB203 (Coherus, Phase 3), SB15 (Biogen-Samsung, Phase 3), ALT-L9 (Alteogen, pre-clinical), SCD411 (Sam Chun Dang, Phase 3), AVT06 (Alvotech, Phase 3), CT-P42 (Celltrion, Phase 3), SOK583A1 (Sandoz-Hexal), and M710/MYL-1701P (Mylan-Momenta, pending) are among the candidates [23], [26].
The details are as follows in paragraph form:
Initially sold by Johnson & Johnson under the brand name REMICADE, infliximab has a number of biosimilars, such as INFLECTRA (Pfizer), RENFLEXIS (Organon), AVSOLA (Amgen), and NI-071 (Nichi-Iko), which is presently undergoing Phase 3. Biosimilars of etanercept, which Amgen sells under the brand name ENBREL, include ERELZI (Sandoz), ETICOVO (Samsung), and YLB113 (Lupin, Phase 3). Several biosimilars of adalimumab, which AbbVie markets as HUMIRA, including AMJEVITA (Amgen), CYLTEZO (BI), HULIO (Viatris), HYRIMOZ (Sandoz), ABRILADA (Pfizer), YUSIMRY (Coherus), HADLIMA (Organon), IDACIO (Fresenius), YUFLYMA (Celltrion), and AVT02 (Alvotech-Teva, pending approval) [10], [23].
Natalizumab has a biosimilar called TYRUKO (Sandoz), which is sold by Biogen under the brand name TYSABRI. Tocilizumab, which Genentech sells under the brand name ACTEMRA, has biosimilars such as DRL_TC (Dr. Reddy's, Phase 3), Tyenne (Fresenius, pending), TOFIDENCE (Biogen), and CT-P47 (Celltrion, Phase 3). WEZLANA (Amgen) is a biosimilar of ustekinumab, which Johnson & Johnson markets as STELARA. Xcimzane (Xbrane-Biogen, preclinical) is a biosimilar candidate for certolizumab, which UCB sells as CIMZIA. AVT05 (Alvotech, Phase 3) and BAT2506 (Bio-Thera, Phase 3) are biosimilars of golimumab, which was first marketed as SIMPONI by J&J [10]. Biosimilars of eculizumab, which Alexion markets as SOLIRIS, include SB12 (Samsung Bioepis, Phase 3) and ABP 959 (Amgen, pending). Biosimilar prospects for omalizumab, which Alexion sells under the brand name XOLAIR, include CT-P39 (Celltrion, Phase 3), BP11 (Aurobindo, Phase 3), and TEV-45779 (Teva, Phase 3). In addition to the biosimilars TRUXIMA (Teva), RUXIENCE (Pfizer), and RIABNI (Amgen), Rituximab, formerly known as RITUXAN by Genentech, has numerous more under development, including DRL RI (Dr. Reddy's, Phase 3), SAIT101 (AZ-Archigen, Phase 3), and MABIONCD20 (Mabion, Phase 3). [10], [26]. Bevacizumab, which Genentech sells under the brand name AVASTIN, has several biosimilars, such as MVASI (Amgen), ZIRABEV (Pfizer), ALYMSYS (Amneal), VEGZELMA (Celltrion), and AVZIVI (Sandoz). Other potential candidates include SB8 (Organon-Samsung, pending), FKB238 (AZ-Centus, pending), TX16 (Tanvex, Phase 1), and ABEVMY (Mylan-Biocon, pending). The Genentech-marketed drug trastuzumab, also known as HERCEPTIN, has biosimilars KANJINTI (Amgen), OGIVRI (Mylan), TRAZIMERA (Pfizer), and HERZUMA (Teva). Other pending or investigational candidates include TX05 (Tanvex), EG12014 (Sandoz), HD201 (Prestige Bio, Phase 3), and Zercepac (Accord, pending) [10].
Regulatory Aspects
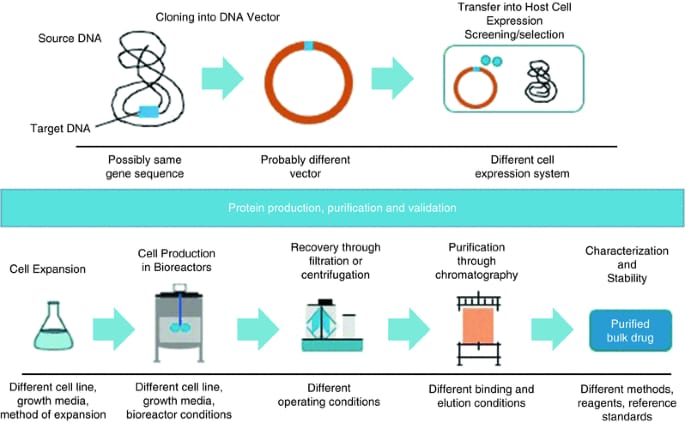
Most people agree that the generic strategy for chemically synthesized small molecules is inappropriate for biosimilars. A special rule based on biosimilarity demonstration concerning quality, safety, and efficacy issues in relation to a reference product governs the approval of "copies" of biologics known as biosimilars. Regulatory bodies do not agree on the term "biosimilar," and each one uses a different definition [24]. The fundamental ideas guiding regulatory criteria are quite similar, even though there are minor variations in the range of rules, reference product attributes, and datasets needed for approval across various regions [7]. Beginning with first-generation biologics (somatropin) and progressing to complex compounds like erythropoietin and monoclonal antibodies (mAbs) (infliximab), the EU was a pioneer in the establishment of regulatory aspects for biosimilars as well as in granting marketing authorization for them. In 2005, the European Medicines Agency (EMA) released a basic framework guideline for biosimilars, outlining the biosimilarity principles that form the cornerstone of most subsequent guidelines [2], [10]. The Committee for Medicinal Products for Human Use (CHMP) accepted a revised draft guideline in October 2014 after it was issued in 2013 in response to technical improvements and application review experience [23]. The reference product for proving biosimilarity is still the one that has received European Economic Area (EEA) approval. According to the updated guideline, some clinical studies and in vivo non-clinical studies could be carried out using a non-EEA-approved reference product that provides justification and bridging studies in order to encourage the global development of biosimilars and prevent the duplication of clinical trials. For a product approved by a regulatory body with comparable scientific and regulatory criteria to EMA, this citation is appropriate. [7]
Biosimilars: an economic boon
The main benefits of biosimilars are their lower cost, ability to compete with the original innovator biologic, and potential improvement in immunologic profile. Therefore, understanding their cost-effectiveness and the financial benefit they provide is essential [11].
According to a report by the Novartis-funded Drug Discovery and Development (IMS) Institute, the eight best-selling biologics in the US and five European nations (Germany, France, Italy, Spain, and the UK) will cost a combined $225 billion between 2016 and 2020 if biosimilars do not compete. The total savings in the presence of biosimilars might be between $45 and $90 billion. 10. By 2024, the US-FDA estimates that biosimilars will save $250 billion [1], [14].
CHALLENGES AND OTHER ISSUES WITH BIOSIMILARS
Biological drift and biological divergence
In contrast to tiny molecules, biological agents are naturally variable and extremely susceptible to modifications in the production process. Clinically significant discrepancies in safety, effectiveness, and immunogenicity may arise from known sponsor modifications to enhance production quality, efficiency, and convenience as well as from unknown or unexpected (drift) changes in the manufacturing process system. A process known as biological divergence can develop when two products that were originally very similar lose a significant amount of their similarity due to unchecked drift and/or other changes that accumulate over time. The comparator utilized in the comparability exercise may eventually alter as a result, and since there is no discernible pairwise difference, biosimilarity may be demonstrated. Strong quality characteristic control systems and clearly defined standards can help to lessen this [18].
Pharmacovigilance
All potential variations between the reference product and the suggested biosimilar cannot be explained by the comparability exercise required to prove biosimilarity. The biosimilar may have a different safety profile in terms of the frequency and severity of adverse effects, even though it may show comparable efficacy. The biosimilar product, the biologic's originator, and the patient's characteristics may also alter over time. Therefore, there is a greater need for ongoing evaluation of the biologicals' risk-benefit ratio, particularly for second-generation biosimilars. A strong post-marketing surveillance system and a risk management strategy are primarily required by regulatory bodies, especially when it comes to the immunogenicity issue. This covers strategies including creating patient registries, tracking prescription events, improving adverse event reporting, and more [15].
Substitution
Pharmacists can dispense generic medications instead of innovator goods ordered by doctors without the treating physician's knowledge or approval thanks to automatic substitution. Automatic substitution is appropriate and can result in cost savings for most small-molecule generics. The majority of small-molecule generics can save money by using automatic replacement, which is acceptable. However, automatic substitution can compromise safety and pharmacovigilance efforts and is not usually advised. Modified-release theophylline and calcium channel blockers are examples of medications having a limited therapeutic index for which automatic substitution may not be suitable. It is unrealistic to expect that the generic version of these medications will have the same risk/benefit profile as the original since the concentrations needed to generate a therapeutic effect and those linked to toxicity differ too little [15].
Nomenclature and identification
The specific biologic medication associated with the adverse event must be easily recognized and reported by sponsors, regulatory bodies, and other medical professionals, including prescribers. The problem of identification and traceability is more difficult in the case of biologicals, though, because the medications may vary over time, leading to variations between batches and manufacturers of the same biological drug, which is made more difficult by interchangeability. Because biosimilar medications are comparable to the reference product rather than identical, they cannot be categorized as generic medications. Because of this, biosimilar medications cannot be referred to by the same non-proprietary name as generic small-molecule medications. It is crucial that patients, healthcare providers, and regulators can recognize the biosimilar being used and distinguish it from other biosimilars on the market.
The majority of regulatory bodies suggest adding a special suffix to the biological product's INN name. For instance, infliximab-dyyb INFLECTRA and filgrastim-sndz ZARXIO. The trade name is used by several agencies together with Greek letter suffixes and the INN (SANDOZ filgrastim). 20 FDA regulations require that the biosimilar's proprietary name be used for labeling and identification [13], [15].
Future evaluation of biosimilars
Healthcare is now more economical and accessible thanks to biosimilars, but they must be profitable.
The availability of biosimilars has reduced healthcare expenditures and improved patient access. Nevertheless, a lot of patients who could use biologics find it difficult to obtain timely and reasonably priced access. This is true in the developed markets of the US and Europe, and access is considerably more restricted in many other nations [1], [2].
The creation of biosimilars is still costly and can take up to US$300 million and nine years per biosimilar, even though it is anticipated that they will be less expensive and develop more quickly than a completely new originator treatment, whether it be a chemical or biological drug. Comparative clinical trials are the most expensive to develop since they need to buy the reference biologic and recruit enough patients to reach predetermined goals. Developing and launching biosimilars to many originator biologics is becoming more and more difficult for sponsors due to the time and expense involved. The goal is to find process efficiencies that take into account the knowledge that all parties involved have accumulated over the previous 20 years of biosimilar development [8]. The common objectives in healthcare are still the same: increased access and affordability on a global scale through competition without sacrificing efficacy, safety, or quality [11].
It has been estimated that there may not be any biosimilar competitors for roughly half of the biological medications that are set to go off patent in the next ten years. This might be partly because, under the existing development paradigm, their market size is too tiny to justify the expense of developing a biosimilar [10].
Many parties have recognized the potential to expedite the development of biosimilars. At the same time, there will probably be a rise in demand for biologics at lower costs in all developed and developing nations. Only if biosimilars can be created and produced more effectively will there be viable and sustainable competition in every location [17].
CONCLUSION
Biosimilars provide affordable substitutes for expensive biologic medicines and are a significant development in contemporary healthcare. Biosimilars provide prospects for greater market competition, wider patient access, and substantial global healthcare savings as original biologics' patents expire. To maintain safety and effectiveness in spite of these advantages, issues including biological variability, regulatory complexity, pharmacovigilance requirements, and naming conventions must be resolved. Investment in medications with smaller market sizes is frequently discouraged by the time and resource commitment required to create biosimilars. Nevertheless, new technology and changing regulatory frameworks offer encouraging opportunities to expedite the development of biosimilars, lower costs, and increase their accessibility worldwide. Stakeholders must work together to foster innovation, improve regulatory harmonization, and raise patient and provider knowledge in order to attain sustainable access. In the end, biosimilars are expected to become more and more important in enhancing healthcare affordability without sacrificing quality.
REFERENCES



Dinesh Vhanale, Deepti Bandawane, Pratiksha Hajare, Biosimilars: Opportunities, Challenges, and the Future of Affordable Biologics, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 3, 2468-2476. https://doi.org/10.5281/zenodo.19146077
 10.5281/zenodo.19146077
10.5281/zenodo.19146077
