Department of Pharmaceutics, College of Pharmaceutical Sciences, Govt. Medical College, Kozhikode
Bilosomes are bile salt (BS) stabilized vesicular nanocarriers that have emerged as promising platforms for oral drug and vaccine delivery. Incorporation of bile salts into lipid bilayers enhances vesicle stability against gastrointestinal degradation and promotes intestinal permeation. This review comprehensively discusses the structural features, composition, and preparation methods of bilosomes, including thin-film hydration, reverse-phase evaporation, and ethanol injection techniques etc. The mechanisms underlying vesicular transport, particularly lymphatic uptake and M cell mediated transcytosis, are highlighted to explain their ability to bypass first pass hepatic metabolism and enhance systemic bioavailability. Critical characterization parameters such as vesicle size, zeta potential, entrapment efficiency, morphology, in vitro release, and stability studies are systematically outlined. Furthermore, recent pharmaceutical applications of bilosomes in oral delivery of peptides, proteins, vaccines, and poorly bioavailable drugs are examined. Owing to their improved stability, mucosal immunogenicity, and enhanced therapeutic performance, bilosomes represent a versatile and efficient vesicular delivery system with significant potential in modern drug delivery research.
There is different route of drug delivery systems, among these oral routes considered to be most popular. However, many commercially available drugs have significant first-pass metabolism, restricted membrane permeability, and poor water solubility makes low and inconsistent oral bioavailability. Many formulation techniques have been developed to solve these issues. When compared to conventional dosage forms, vesicular drug delivery methods have drawn a lot of interest for oral administration because of their capacity to improve drug stability, membrane permeability, and bioavailability.[1] Numerous vesicular systems such as Liposomes[2], Noisomes[3], Bilosomes[4], Transferosomes[5] and Cubosomes[6] etc have been investigated in pharmaceutical formulations.
The term "bilosomes" was first coined in 2001 by Conacher et al. during their investigation on oral immunization using on influenza subunit vaccine, a synthetic measles peptide and bovine serum albumin as a model antigen. Bile salts were added to stabilize in this formulation after successful encapsulation of the antigen within the lipid based non-ionic surfactant vesicles.[7]
Bilosomes made up of amphiphilic bile salts, non-ionic surfactants and cholesterol are flexible and deformable. Due to the presence of bile salt in the vesicle membrane, which improves penetration and increases the effectiveness of oral administration. Bilosomes have significant benefits over conventional vesicular systems like liposomes and noisomes. These advantages include the ability to withstand enzymatic degradation in the gastrointestinal tract. Bile salt imparts a negative charge to the vesicle, enhancing their storage stability and facilitating drug uptake through M cells in Peyer’s patches. This mechanism helps bypass hepatic first pass metabolism and support drug transport via the intestinal lymphatic system. Consequently reduce dosing frequency leads to better patient compliance.[8] The Hydrophile–lipophile balance (HLB), chemical structure, and critical packing parameter (CPP) of the surfactants are influencing factors for vesicle formation. The Tween and Span series have shown excellent entrapment efficiency for hydrophilic medicines, especially when combined with cholesterol.[9]
COMPOSITION OF BILOSOME
1) Bile salt
The liver produces endogenous amphiphilic steroidal molecules called bile salts, which are then released into the digestive system. They have unique hydrophilic (hydroxyl groups) and hydrophobic (methyl groups) surfaces due to their facial (planar) polarity, which facilitates effective interaction with biological membranes. Bile salts function as natural detergents in the digestive system, combining with phospholipids and dietary lipids to generate mixed micelles that improve the solubility of medications that are not very soluble in water and make it easier for the body to absorb them. There are various bile salts available for the formation of bilosomes such as Sodium deoxycholate (SDC), Sodium cholate (SC), Sodium glycocholate (SGC), Sodium taurocholate (STC), Sodium Tauro deoxycholate (STDC)etc. Among these Sodium deoxy cholate is the most commonly used.[10]
2) Non-ionic surfactants
Non-ionic surfactants are the most commonly utilized surface-active agents in vesicular drug delivery systems since they are more stable, biocompatible and less toxic than ionic surfactants. In addition to maintaining pH close to physiological, they are less irritating and haemolytic and serve as permeability enhancers, emulsifiers, solubilizers, and wetting agents.[11] Span and Tween series of non-ionic surfactants are frequently utilized in bilosome formulations because of their superior stability, low toxicity, and biocompatibility.[12]
3) Lipid components[13]
(a) Phospholipid
Phospholipids have a hydrophilic phosphate group and a hydrophobic acyl chain in their structure; they are known as amphipathic compounds. As a result, they have outstanding self-assembling, biocompatible, and structural qualities. They thereby exhibit superior wetting, emulsifying, self-assembling, and biocompatibility qualities.
(b) Cholesterol
The physicochemical characteristics and stability of bilosomes are significantly influenced by cholesterol. It is an amphiphilic molecule that integrates into the bilayer membrane with its hydrophobic aliphatic chain aligned with the lipid acyl chains and its hydroxyl group orientated toward the aqueous phase. In order to contribute to overall vesicle stability, this configuration increases membrane rigidity, decreases permeability, improves structural integrity, and minimizes leaking of entrapped hydrophilic medicines.
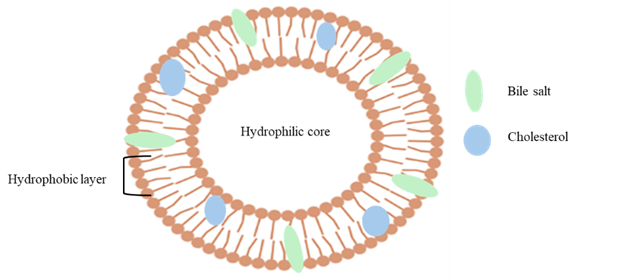
Figure1: Showing a Bilosome vesicle structure
COMPARISON WITH CONVENTIONAL VESICULAR SYSTEMS
Table:1 Showing comparison of different vesicular systems
|
Characterisation parameters |
Liposomes[2] |
Niosomes[3] |
Bilosomes[13] |
|
Size |
50nm-150nm |
unilamellar vesicles (0.025-0.05µm) multilamellar vesicles(>0.05µm) large unilamellar vesicles(>0.10µm) |
50nm-200nm |
|
Composition |
phospholipid, charge inducer(surfactant) |
non-ionic surfactants, cholesterol |
non-ionic surfactant, bile salt |
|
Stability in GIT |
unstable |
unstable |
stable |
MECHANISM OF BILOSOME
Drugs that are absorbed from the intestine either reach the systemic circulation through the intestinal lymphatic system or through the portal vein (undergoes first-pass metabolism). Bilosomes are preferentially absorbed via the lymphatic route because of the presence of bile salt and lipid-rich nature. Higher systemic drug concentrations and better therapeutic efficacy arise from this lymphatic passage leads to prevention of first-pass metabolism, particularly for medications that are prone to quick hepatic metabolism.[4]
MODIFICATIONS OF BILOSOME
Probilosomes were improve oral bioavailability and shield acid-labile medications from deterioration in the stomach environment. Bile salt incorporation into the vesicular structure facilitates better intestinal absorption and prevents the stomach from releasing drugs early.[14] Polyethylene glycol polymer used to modify the bilosome surface to enhance drug loading and extend drug release is known as Pegylated bilosomes. The polymer coating increases circulation time, lowers reticuloendothelial system clearance, and improves mucoadhesion leads to enhance therapeutic efficacy.[15] Chitosan, a naturally occurring cationic polymer with bio adhesive, biodegradable, and biocompatible qualities is used to modify the surface of bilosomes. Through electrostatic interaction of positively charged chitosan with negatively charged mucosal membrane enhance penetration by promoting mucoadhesion and opening tight epithelial junctions. Additionally, the coating creates a barrier around the vesicles which slows down drug release and increase the stability. In addition, chitosan improves cellular absorption and imparts antibacterial activity, which results in increased biological efficacy as compared to the uncoated formulation.[16] Carbohydrates like dextrose to modify the surface of bilosomes. In order to facilitate lectin–carbohydrate interactions with receptors expressed on liver and spleen cells, the carbohydrate moiety functions as a targeting ligand. This surface functionalization improves retention at the target site after oral delivery, strengthens stability against gastrointestinal degradation, and improves organ-specific accumulation.[17] Triblock copolymer (Pluronic P123) and a bile salt (sodium cholate hydrate) were used to surface modify phosphatidylcholine/cholesterol-based bilosomes. The alteration enhanced colloidal stability and made it possible to effectively encapsulate both hydrophilic and hydrophobic medications gives stable and adaptable drug delivery method.[18] Bilosomes are effective oral vaccine delivery vesicle that shield encapsulated antigens from premature release and degradation in the harsh gastrointestinal environment by encapsulating them in a lipid bilayer. The effective activation of the mucosal immune system depends on the absorption of antigens by Peyer's patches particularly by M cells. Bilosomes are internalized by M cells and then transferred to underlying antigen-presenting cells (APCs) which process and present the antigen to B and T cells therefore triggering antigen-specific systemic and mucosal immune responses.[7]
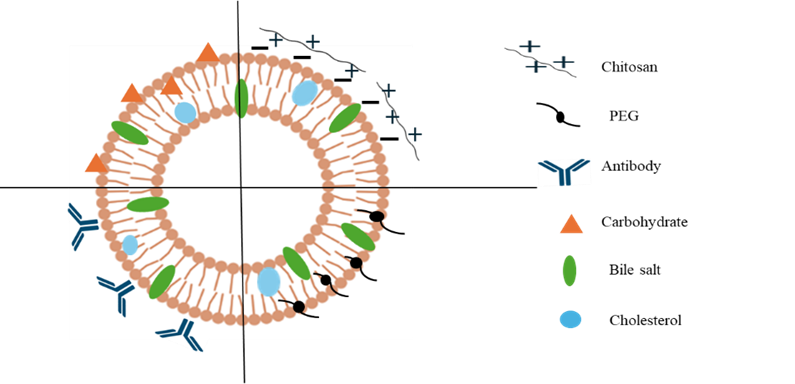
Figure 2: Showing surface modified bilosome vesicle
PREPARATIONS OF BILOSOME
followings are different techniques available to prepare bilosomes
1) Thin film hydration method
2) Reverse phase hydration method
3) Solvent evaporation method
4) Ethanol injection method
5) Melt method
6) Probe sonication method
1)Thin film hydration method
Among the different techniques thin film hydration method seen as the most common method employed. By using rotary evaporator, made a thin film of lipid and then add the aqueous solution to it. Lipid components like phospholipid, cholesterol and drug were dissolved in appropriate organic solvents such as methanol or chloroform or its mixture generally in round bottom flask. This organic phase then subjected to evaporation by using rotary evaporator under reduced pressure to form thin lipid film on the inner wall. Aqueous solution of bile salt which is prepared either with dissolving of bile salt with saline phosphate buffer or dissolved with distilled water or with any other solution is added to this thin lipid film with continuous agitation to get bilosomal dispersion. The resultant mixture then subjected to sonication with defined cycles and intermittent intervals for the reduction of particle size of the vesicles. Finally prepared bilosomal dispersion was kept in refrigerator for further characterisation.[19,20]
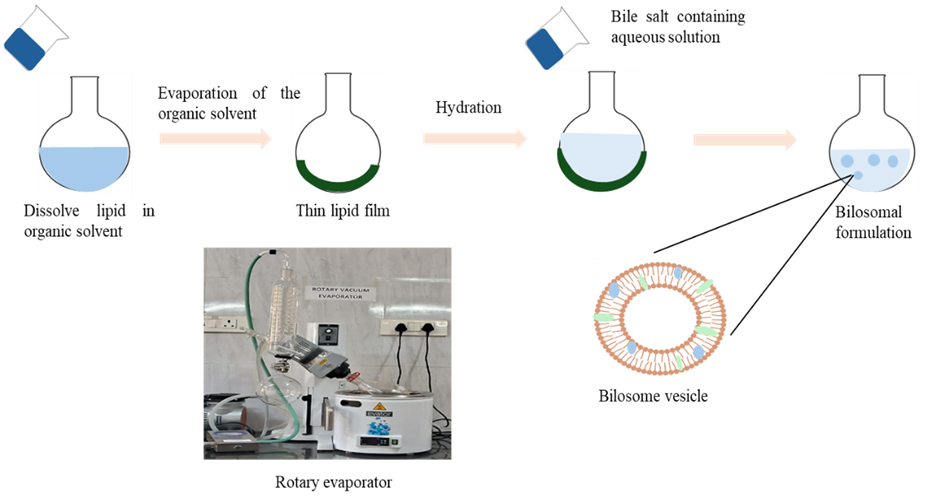
Figure 3: Showing preparation by using thin film hydration technique
2)Reverse phase hydration method
Mixture of surfactants (mainly span 60 and tween 60) and cholesterol were added to the round bottom flask with adapter containing organic solvent (may be ethanol, chloroform, diethyl ether etc.). Aqueous solution of bile salt and drug were prepared. These two phases are then mixed using ultrasonic bath to get bilosomal emulsion which is then subjected to rotary evaporator for drying. Rehydrated with solvent (distilled water, deionised water). the resultant dispersion heated with water bath and then sonicated for reduction of vesicular size. Final dispersion store at 4?c for the evaluation.[21]
3)Solvent evaporation method
Appropriate amount of cholesterol and drug were dissolved in organic solvent (ethanol, chloroform-methanol mixture etc.). Aqueous solution of bile salt was prepared. Organic phase added to the aqueous phase upon heating with continuous agitation until the complete evaporation of organic solvent from the resultant mixture occur. Then subjected to bath sonication for to get the reduced particle size of the vesicles.[16,22]
4)Ethanol injection method
Lipid, non-ionic surfactant and drug were dissolved in preheated ethanol and aqueous solution of bile salt were also preheated separately at the same temperature. The ethanol solution injected to the aqueous solution which taken in a beaker at controlled rate using syringe under continuous magnetic stirring. Resultant mixture became turbid. Stirring continuous up to the complete evaporation of organic solvent then subjected to the sonication for reduction of particle size. store at 4?c.[23,24]
5)Melt method
Phospholipid, cholesterol, diacetyl phosphate was taken in appropriate molar ratio which is then subjected to heating. Bile salt and drug were dissolved in buffer and vortexed for 2 minutes. The un entrapped vesicles were removed by centrifugation.[25]
6)Probe sonication method
Appropriate amount of non-ionic surfactant, cholesterol, bile salt and drug were taken in a beaker. Added distilled water to the above mixture and then homogenized by using homogenizer at 3000 rpm for 5 minutes. Then after probe sonication was done for 5 minutes. Resultant mixture turns into milky dispersion .kept in refrigerator for further evaluation.[26]
CHARACTERISATION OF BILOSOME
Entrapment efficiency
Particle size, Zeta potential, Polydispersity index
Vesicular morphology
Invitro drug release
Ex vivo studies
In vivo pharmacokinetic study
Physical stability
Solid state characterisation like DSC and FTIR
Release kinetics
Statistical analysis
1)Entrapment efficiency
The ultra-centrifugation technique was used to determine the entrapment efficiency of bilosomes in an indirect manner. The bilosome formulation was centrifuged for 90 minutes at 4°C and 14,000 rpm using a cooling centrifuge. After the supernatant was removed, a UV spectrophotometer (Shimadzu, UV-2401 PC, Kyoto, Japan) was used to determine the presence of the free drug was present. To prevent any interference, the regression equation of the standard curve created in phosphate buffer was used to calculate the entrapped drug concentration using a blank. Encapsulating efficiency was calculated by the following equation.[27]
EE%= Theoratical amount of the drug-Actual amount of drug presentTheoratical amount of the drug×100
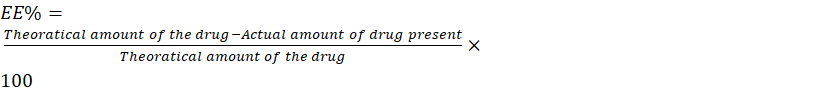
2)Particle size, Zeta potential, Polydispersity index
Zeta sizer(Malvern nano zetasizer,uk)used to measure the particle size, zeta potential and polydispersity index(PDI) of the bilosome dispersion by diluting with deionized water and measurement taken in triplicate.[28]
3)Vesicular morphology
For the morphological examination, the optimal bilosome formulation was selected. On a carbon-coated grid, one drop of freshly made, diluted bilosomal suspension was put. It was then allowed to dry for five minutes at room temperature. To ensure adequate staining absorption, samples were treated with 1% (w/v) phosphotungstic acid as a negative stain. After two minutes, the surplus liquid was removed using filter paper. The samples were scanned using a TEM (transmission electron microscopy).[29]
4)In vitro drug release
Using the dialysis membrane diffusion technique in a shaking water bath, invitro release of optimized vesicular formulation was evaluated. Prehydrated dialysis bags were filled with measured volume of the formulation and both ends were sealed. The dialysis bags were kept at 37±0.5?c with constant shaking at 100 rpm while submerged in phosphate buffer that contained appropriate surfactant to maintain sink conditions. Aliquots were taken out replaced with equivalent volume of fresh medium at specified intervals; each sample was examined using spectrophotometry. plotting the cumulative amount of drug release against time and mean ±SD was used to express the result.[30]
5)Ex vivo permeation method
Ex vivo performed in male wilstar rats using noninverted gut sac method. A section of the small intestine was carefully separated after the animals were anesthetized. In order to create a sac, one end of the intestinal segment was ligated, filled with the test formulation, and firmly knotted at the other end. With constant stirring and aeration, each sac was submerged in a suitable physiological buffer that was kept at 37 °C. Fresh media was added in to the receptor medium with equivalent amount of sample withdrawn at predetermined intervals. The drug content measured using UV spectrophotometry. The total drug penetration per unit surface area was computed. The findings were presented as mean ± SD, and each experiment was run in triplicate.[31]
6)In vivo pharmacokinetic activity
Healthy adult female Wistar rats were used in the investigation. The animals were split into two equal groups with the acceptance of Animal Ethics Committee. Rats were fasted overnight and continued to be fasted for two hours after the dose was given for the oral absorption investigation. An equivalent oral dose of the pure medication was given to the second group, whereas one group received a single oral dose of the improved freeze-dried bilosomal formulation. Using an oral feeding tube, the formulations were given orally as aqueous suspensions. During a 24-hour period, blood samples were obtained by orbital sinus puncture while under anaesthesia at prearranged intervals. Centrifugation was used to separate the plasma, which was then kept at a low temperature pending additional pharmacokinetic analysis.[32]
7)Stability studies
A stability investigation was conducted at two distinct temperatures: 4? C and 25? C with 60% relative humidity. For three months, the sample was kept in a chamber after being sealed in borosilicate glass vials. The sample was taken out at specific times (0, 30, 60, 90, and 180 days) and the Vesicular Size and EE was examined.[15]
8)Solid state characterisation.[33]
(a) DSC
Differential scanning calorimetry (DSC) was used to examine the thermal properties of both the optimized bilosomal formulation and the pure medication. To evaluate potential thermal transitions and drug–excipient interactions, precisely weighed samples were put in sealed aluminium pans and heated over a predetermined temperature range at a regulated heating rate.
(b)FTIR
Both pure drug and optimized bilosomes analysed by using FTIR spectrometer (SHIMADZU FTIR 8400S)
9)Release kinetics
different kinetic studies like Zero order kinetics, First order kinetics, Higuchi method, Korsmeyer Peppas model and Hixson-Crowell model were employed.[34]
10)Statistical analysis
Result expressed as Mean±SD and statistical analysis was carried out using GraphPad prism in which p value<0.05 was considered as statistically significant.[35]
ORAL IMMUNIZATION OF BILOSOME
Specialized epithelial cells known as microfold (M) cells are seen in the Peyer's patches of the small intestine within GALT (Gut associated lymphoid tissue) and in MALT (Mucosa associated lymphoid tissue) of other gastrointestinal areas. By moving bacteria and particles from the gut lumen over the epithelium to the lamina propria, they initiate mucosal immune responses. In contrast to normal enterocytes, M cells have an intraepithelial pocket with macrophages and lymphocytes that engage with transcytosed antigens. They also lack a brush boundary. Bilosomes loaded with antigens are transported to this region, where immune cells initiate mucosal and systemic immunity. M cells also release IL-1, which gives T and B cells in the Peyer's patches milieu costimulatory cytokine signals.[36] Oral bilosome-based vaccinations provide systemic and mucosal immune responses similar to parenteral vaccines without any discernible harm, according to preclinical research in BALB/c mice. Unlike traditional intramuscular alum-adsorbed vaccines, influenza, tetanus toxoid (TT), hepatitis B surface antigen (HBsAg), and diphtheria toxoid (DTX)-loaded bilosomes produced substantial serum IgG responses in addition to eliciting significant mucosal IgA. Bigger vesicles improved Th1 responses (IgG2a, IFN-γ), while bilosomes enhanced viral clearance, mucosal penetration, antigen protection, and Peyer's patch uptake. While subcutaneous live vaccines frequently resulted in higher neutralizing antibody titres, use of adjuvants like recombinant Bac-VP1 or CTB enhanced immune responses further.[37]
APPLICATIONS
Table 2: Applications of bilosome vesicular drug delivery
|
Active agent |
Delivery route |
Formulation technique |
Bilosome composition |
Reference |
|
Insulin |
Oral |
Reversed phase evaporation method |
SPC:BS (4:1 ratio) RhIN (Recombinant human insulin- 4 mg/ml) |
[38] |
|
Eprosartan mesylate |
Oral |
Thin film hydration method |
drug(30mg) SPC in chloroform: methanol (2/1 v/v) SDC in phosphate buffer |
[1] |
|
Fenofibrate |
Oral |
Dry film dispersion method |
drug(10mg), SPC (300mg) dissolved in diethyl ether, SDC in 10ml phosphate buffer |
[39] |
|
Cyclosporin |
Oral |
Dry film dispersion method |
SPC, SDC, CyA dissolves in chloroform: methanol (9/1, v/v) |
[40] |
|
Nisoldipine |
Oral |
Thin film hydration method |
drug(10mg), CH (Cholestrol-80mg) Span 60 (240mg) in organic phase. SDC (5,10,20mg), SGC(5,10,20mg) |
[8] |
|
Resveratrol |
Oral |
Thin film hydration method |
SPC: SDC:CH (4:1:0/4:1:1 ratio) drug(5,10,20,30mg/ml) |
[28] |
|
silymarin |
Peroral |
Thin film hydration method |
Tween 20(100,200,300mg) and Syl in methanol, Span 60and CH (10-20mg) in chloroform, SDC (50-150mg) in phosphate buffer |
[17] |
|
Linagliptin |
Oral |
Solvent evaporation method |
CH (2.5,5,7.5mg) and drug(5mg) in ethanol, TGPS (Tocopherol polyethylene glycol succinate) and SDC (10,20,30mg) in aqueous solution |
[22] |
|
Avenanthramide |
Oral |
Thin film hydration method |
L-αphosphatidyl choline(10mg), CH (1,2,4mg/ml) drug(1mg/ml) in ethanol BS (1,2,4mg/ml) in distilled water |
[41] |
|
Hesperetin |
Oral |
Thin film hydration method |
Span 60(100,300,500mg), CH (0.5,2.75,5%) PEG, BS (0.1,0.3,0.5mg) in chloroform: methanol (1:1, v/v) |
[42] |
|
Levofloxacin and Doxycycline |
Oral |
Melt method |
Phospholipid, CH, diacyl phosphate (5:4:1 M ratio), SDC in 0.025 M in carbonate buffer |
[25] |
|
Lacidipine |
Oral |
Ethanol injection |
Drug, Span 60(75,187.5,300mg), CH (25,47.5,70mg) in ethanol, SDC(5,10,15mg) in aqueous solution |
[23] |
|
Dasatinib |
Oral |
Reverse phase evaporation method |
CH, SA (Tween 60, Span 60), drug(20mg) in ethanol SDC(5,10,15mg) and soluplus(50,75,100mg) in aqueous solution |
[43] |
|
Trametinib |
Oral |
Thin film hydration method |
drug(2.5mg) CH (10mg) and STC (10-30mg) |
[44] |
|
Berberine |
Oral |
Thin film hydration method |
SPC (0.03,0.06,0.045 M), berberine(10mg) and CH (15,30,22.5mg) in chloroform: methanol (2:1ratio) SDC (15,30,22.5mg) in phosphate buffer |
[45] |
|
Ondansetron |
Transdermal |
Thin film hydration method |
Span60:CH (7:0-7:3) and SDC (2.5,5mg) |
[46] |
|
Metformin |
Transdermal |
Solvent evaporation method |
Drug(600mg), CH (14mg) in methanol: methylene chloride (1:2), SA (Span 40, Span 60) Edge activator (STC-8,14mg, SC-8,14mg, SDC-8,14mg) |
[27] |
|
Simvastatin |
Transdermal |
Thin film hydration method |
SPC (1,2,3M) Span 60(30,45,60mg) in chloroform: methanol (1;1, v/v) SDC (10,20,30mg) in phosphate buffer |
[19] |
|
Dapsone |
Cutaneous delivery |
Thin film hydration method |
Drug(20mg), Span 60:CH (5:1,10:1M ratio) in chloroform, SDC/SC/STC (0.25,0.5 mg) in distilled water |
[47] |
|
Valsartan |
Transdermal |
Thin film hydration method |
Drug(40mg), Span 20,40,60:CH 60(1:1,2:8,8:2mg) in chloroform: methanol (7:3) SDC(5,10,20mg) in distilled water |
[48] |
|
Tenoxicam |
Transdermal |
Thin film hydration method |
drug(20mg), SA (Surfactant-span 40,60,80):CH 5:1and 5:3 ratio dissolved in chloroform, SDC (0.5,0.25mg) in distilled water |
[49] |
|
Terbutaline sulphate |
Transdermal |
Thin film hydration method |
SPC(3%,4%,5%w/v) CH (20mg) SDC(5,10,15%w/v), chitosan (0.3,0.15%w/v) |
[29] |
|
Dronedarone |
Transdermal |
Ethanol injection method |
Span 40, CH, Clove oil and drug in ethanol SDC and tween 60and 80 in aqueous solution |
[50] |
|
Meloxicam |
Transdermal |
Probe sonication method |
Span 60(420mg) drug(5,10,15mg) and CH (60,80,300mg), SDC (5,10,15mg) |
[26] |
|
Tenoxicam |
Transdermal |
Thin film hydration method |
drug(20mg), Span (40,60,80):CH (5:1,5:3) in chloroform and SDC (0.25,0.5M) in distilled water |
[49] |
|
Resveratrol |
Intranasal |
Thin film hydration method |
CH; Span60(1:1,1:2,2:1), drug(10mg) in chloroform: ethanol: methanol (7:1:2) SDC (10mg) |
[28] |
|
Zolmitriptan |
Intranasal |
Thin film hydration method |
Drug(20mg), CH: Span in different molar ratio. SDC (5,10,15) |
[51] |
|
Glibenclamide |
Intranasal |
Thin film hydration method |
Drug(3mg), CH, Span 40(1:1,1:5,1:9ratio) SDC(5,15,25mg) |
[52] |
|
Moxifloxacin |
Ocular |
Thin film hydration method |
Span 60(30,45,60), Cremophor EL (7,11,15) in chloroform: methanol (1:1), SDC (15,22.5,30mg) in aqueous solution |
[20] |
|
Acetazolamide |
Ocular |
Thin film hydration method |
Drug, Span 60, CH in acetone: methanol (1:1, v/v), SDC, SC, STC, STGC (1:1:0:1,1:1:0:2M ratio) |
[53] |
|
Tacrolimus |
Ocular |
Thin film hydration method |
Soyabean phosphatidyl choline: SDC/SGC/STC (5:1 ratio), drug (5.9%) |
[54] |
|
Daclatasvir and Xanthone |
Respiratory |
Thin film hydration method |
phosphatidyl choline (150mg): CH (3:1 M ratio) dissolved in methylene chloride, xanthone drug (20mg), SDC (15%w/v) in aqueous solution |
[55] |
|
Tetanus |
Toxoid |
Thin film hydration method |
Span 80:CH: SDC (2:1:0.1 M ratio) |
[56] |
CONCLUSION
Bilosomes have emerged as a highly promising and versatile vesicular drug delivery system capable of addressing the major limitations associated with conventional drug and vaccine delivery. The incorporation of bile salt into the vesicular membrane significantly enhances their stability against gastrointestinal degradation, improves membrane permeability, and promotes lymphatic uptake, thereby enhancing bioavailability and minimizing first pass hepatic metabolism. These unique structural and functional properties enable bilosomes to effectively protect encapsulated drugs, improve solubility, and provide controlled and targeted drug release. The composition, preparation methods, transport mechanism, characterization parameters and surface modification strategies of bilosomes, highlighting their crucial role in improving therapeutic performance. Surface engineering approaches such as chitosan coating, PEGylation, and ligand mediated targeting further enhance cellular uptake, mucoadhesion site specific delivery thereby improving therapeutic efficacy and reducing systemic side effects. Moreover, bilosomes have demonstrated significant potential in a delivery of wide range of therapeutic agents, including peptides, proteins, vaccines and poorly soluble drugs via oral, transdermal, ocular, intranasal and other delivery routes. Their ability to induce both systemic and mucosal immune immune responses also makes them particularly valuable for oral vaccine delivery. Despite these encouraging advancements, the clinical translation of bilosomal formulations remains limited due to insufficient large scale manufacturing data, long term stability studies and clinical evaluations. Therefore, future research should focus on optimizing formulation parameters, exploring advanced targeting strategies conducting comprehensive in vivo and clinical studies and establishing scalable production methods. Overall, bilosomes represent a robust efficient and innovative nanocarrier platform with substantial potential to revolutionize drug delivery and contribute to the development of safer, more effective therapeutic systems in modern pharmaceutical formulations.
ACKNOWLEDGMENT
The authors would like to express their sincere gratitude to the Kerala University of Health Sciences for providing the academic support and resources necessary to carry out this study. We extend our heartfelt thanks to the faculty members of the Department of Pharmaceutics for their valuable guidance, constructive suggestions, and continuous encouragement throughout the course of this work. We also acknowledge all researchers and authors whose studies have contributed to the literature reviewed in this article. Finally, we thank our peer and well-wishers for their support during the preparation of this manuscript.
REFERENCES


Greeshma V. P., Dr. Geetha V. S., Afeefa K. A., Kadeeja P. S., Sajin K. C. Bilosomes: From Vesicular Engineering to Mechanistic Insights and Therapeutic Applications., Int. J. of Pharm. Sci., 2026, Vol 4, Issue 3, 624-639. https://doi.org/10.5281/zenodo.18897867
 10.5281/zenodo.18897867
10.5281/zenodo.18897867
