Durgamata Institute Of Pharmacy, Dharmapuri, Parbhani, Maharashtra 431401
Retention mechanisms in biological systems help transport essential organic and inorganic substances into the body and maintain their bioavailability. One of the major reasons for poor bioavailability of drugs is inadequate solubility or permeability. Among these, poor aqueous solubility is the most common problem faced by many drug molecules. With the continuous advancement in pharmaceutical and chemical sciences, the need for developing new drug delivery technologies has increased, and the selection of technology depends on the specific properties of each drug. Many drugs exhibit very low water solubility, which limits their dissolution rate in the gastrointestinal tract. Since drug dissolution is a rate-limiting step for absorption, poor solubility directly affects therapeutic effectiveness. The oral route is the most convenient and widely preferred method for drug administration because of its ease of use and patient compliance. According to the Biopharmaceutics Classification System (BCS), drugs are divided into four classes based on solubility and permeability. Solubility-related challenges are mainly observed in BCS Class II and Class IV drugs. To improve the solubility and bioavailability of poorly soluble drugs, various techniques and strategies are applied. These methods include physical and chemical modification of drug substances, particle size reduction, and the use of solubility enhancement technologies. The selection of an appropriate solubility enhancement method depends on several factors such as drug properties, absorption site, and desired dosage form characteristics.
A solvent is generally a liquid that can be either a pure substance or a mixture of two or more liquids capable of dissolving another substance known as a solute. The term insoluble is commonly used for compounds that dissolve very poorly or only slightly in a solvent. Solubility occurs under conditions of dynamic equilibrium, meaning that it results from two simultaneous and opposing processes: dissolution of the solute and recrystallization or phase separation. Solubility equilibrium is achieved when these two processes occur at the same rate under specific conditions [2,3]. In certain situations, the equilibrium solubility may be exceeded, leading to the formation of a supersaturated solution, which is considered metastable. Solubility should not be confused with the ability of a substance to simply melt or chemically degrade. According to the International Union of Pure and Applied Chemistry (IUPAC), solubility is defined as the analytical composition of a saturated solution expressed as the proportion of a specific solute dissolved in a specific solvent.
Solubility is commonly expressed as concentration, such as grams of solute per kilogram of solvent, grams per 100 mL of solvent, molarity, molality, mole fraction, or other related concentration units. For ionic compounds with relatively low solubility, solubility is often described using solubility constants, which represent equilibrium processes. Like other equilibrium constants, temperature significantly affects the numerical value of the solubility constant, while the presence of other dissolved species generally has minimal influence under controlled conditions [5,6,7].
Several theoretical and empirical models are used to predict solubility behavior. The Hansen Solubility Parameters and Hildebrand solubility parameters are commonly applied methods for estimating solubility based on intermolecular interactions. The logarithmic values obtained from these parameters help classify compounds according to their hydrophobic or hydrophilic nature.
Pharmacopoeias such as the United States Pharmacopeia (USP) and British Pharmacopoeia (BP) define solubility using standardized descriptive terms based on the quantity of solvent required to dissolve a specific amount of solute. Wettability studies, including contact angle measurements, indicate improved solubility when drug particles are coated with a hydrophilic layer.
Dissolution rate studies have shown that nanoparticle formulations can significantly enhance drug dissolution. For example, nanoparticles achieved nearly 100% drug dissolution within 5 minutes, whereas the untreated drug material did not dissolve completely even after 120 minutes. These findings demonstrate that nanoparticle technology can substantially improve the dissolution properties of poorly soluble drugs [8,9].
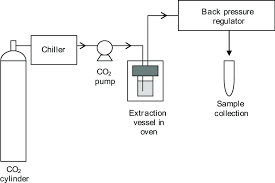
Fig 1: Supercritical liquid Recrystallization.
Supercritical Fluids and Solubility Enhancement
Supercritical fluids (SCFs), such as carbon dioxide, are substances maintained at temperatures and pressures higher than their critical temperature (Tc) and critical pressure. Under these conditions, they exhibit properties of both liquids and gases. Near the critical temperature, SCFs become highly compressible, meaning that small changes in pressure can significantly alter their density and mass transfer properties, which determine their solvent power. When a drug is dissolved in a supercritical fluid, it can recrystallize into particles with much smaller sizes after processing. Carbon dioxide is the most commonly used example because it is safe, inexpensive, and easily removed after processing. Due to high compressibility at critical conditions, SCFs allow control of solvent strength by adjusting pressure. As drugs dissolve in SCFs and later recrystallize, reduced particle size leads to improved dissolution characteristics.
Several pharmaceutical companies, such as Nitra Therapeutics and Lavipharm, specialize in particle engineering technologies using supercritical fluid techniques to reduce particle size and improve drug performance [10,11].
This method involves atomizing aqueous solutions, organic solutions, co-solvent systems, emulsions, or suspensions containing drug substances and excipients directly into compressed gases such as helium, propane, ethane, or cryogenic liquids. Acetonitrile is commonly used as a solvent. This process increases drug loading capacity, reduces drying time, and significantly enhances the dissolution rate compared with supercritical fluid-processed powders.
Role of Surfactants in Solubility Enhancement
Surfactants are highly useful as absorption enhancers because they improve both drug dissolution rate and permeability. They enhance dissolution mainly by improving wetting and allowing dissolution media to penetrate drug particles more effectively.
Sidebar N. et al. [6] studied solubility enhancement of the antimicrobial drug enrofloxacin using various cosolvents and surfactants. The aqueous solubility of enrofloxacin increased up to 26 times [12]. Cosolvents alone produced only a small increase in solubility; however, when combined with buffering systems, a synergistic effect resulted in significant solubility improvement.
Ionic surfactants were found to be more effective solubilizing agents than non-ionic surfactants. Among ionic surfactants, anionic surfactants such as sodium dodecyl sulfate showed higher solubility enhancement compared to cationic surfactants like cetyl trimethyl ammonium bromide, achieving solubility up to 3.8 mg/mL.
Salt Formation
Salt formation improves solubility and dissolution properties compared to the original drug molecule. Generally, a minimum difference of three pKa units between the drug and its counter-ion is required to form stable salts. Alkali metal salts of acidic drugs, such as penicillin, and strong acid salts of basic drugs, such as atropine, show higher water solubility than their parent compounds.
Use of Precipitation Inhibitors
When drug concentration exceeds equilibrium solubility, supersaturation occurs, which may cause drug precipitation or crystallization. This problem can be prevented by using inert polymers such as HPMC, PVP, PVA, and PEG, which act as precipitation inhibitors and maintain drug stability in solution [13].
Co-precipitation Method
In the co-precipitation method, the drug is dissolved in ethanol at room temperature, while a suitable polymer is dissolved in purified water. Different molar ratios of drug and polymer are mixed and stirred for one hour. The solvent is then evaporated, and the resulting mass is ground, passed through sieve No. 80, and stored in desiccators [14].
Precipitation Technique
In this technique, the drug is dissolved in a solvent and then added to an antisolvent to form crystals. The main advantage of this method is the use of simple and low-cost equipment. However, controlling crystal growth is challenging because aggregation may lead to formation of microparticles.
A limitation of this method is that the drug must be soluble in at least one solvent, and that solvent must be miscible with the antisolvent. It is not suitable for drugs poorly soluble in both aqueous and non-aqueous media. Nanosuspensions of diazole and naproxen prepared using this method showed improved dissolution rate and oral bioavailability. Reduction in naproxen particle size resulted in approximately a four-fold increase in absorption rate [15,16].
Spray Drying Technique
In the spray drying method, the drug is first dissolved in a suitable solvent. A required stoichiometric amount of carrier material, such as β-cyclodextrin, is dissolved separately in water. Both solutions are then mixed using sonication or another suitable mixing technique to obtain a clear and uniform solution. The combined drug–polymer solution is processed using a spray dryer, where rapid solvent evaporation takes place. Typically, the drug is dissolved in methanol, while the polymer is dissolved in purified water. These two solutions are mixed to form a homogeneous solution before spray drying. During the drying process, the solvent evaporates quickly, resulting in the formation of a spray-dried drug–polymer complex. The final spray-dried product is usually obtained within 20–30 minutes [17]. Spray drying improves drug solubility by producing fine particles with increased surface area and better dispersion of drug molecules within the carrier material.
Precipitation Method
In the precipitation method, the active drug is dissolved in ethanol at room temperature, while a suitable polymer is dissolved separately in purified water. Different molar ratios of drug and polymer are prepared and mixed individually. The mixture is stirred continuously at room temperature for about one hour, after which the solvent is evaporated. The obtained solid mass is then ground into powder, passed through sieve No. 80, and stored in a desiccator to remove moisture. In this technique, the drug solution is added to an antisolvent, which causes the drug to precipitate in the form of fine crystals [18]. The main advantage of the precipitation method is that it requires simple and low-cost equipment, making it suitable for laboratory-scale preparation. However, careful control is required during crystal formation to prevent aggregation and formation of larger particles. The main advantage of the precipitation method is that it uses simple and low-cost equipment, making it suitable for laboratory and industrial applications. However, one of the major challenges is controlling the growth of drug crystals to prevent the formation of larger microparticles.
A limitation of this technique is that the drug must be soluble in at least one solvent, and this solvent must also be miscible with the antisolvent used during precipitation. Furthermore, this method is not suitable for drugs that are poorly soluble in both aqueous and non-aqueous media.
Nanosuspensions of diazole and naproxen have been successfully prepared using the precipitation method to improve dissolution rate and oral bioavailability. Reduction in particle size of naproxen resulted in an approximately four-fold increase in absorption rate [19].
Spray Drying
In the spray drying technique, the drug is dissolved in a suitable solvent, and a required stoichiometric amount of carrier material, such as β-cyclodextrin, is dissolved separately in an aqueous solution. Both solutions are mixed using sonication or another mixing method to obtain a clear and homogeneous solution.
The drug and polymer solutions are prepared separately, typically by dissolving the drug in methanol and the polymer in purified water. After mixing, the solution is subjected to spray drying, where rapid solvent evaporation occurs. The solvent is removed using an evaporator, and a spray-dried drug–polymer complex is obtained within 20–30 minutes [20,21].
Spray drying enhances solubility by producing fine particles with improved surface area and better dispersion of the drug within the carrier matrix.
Modification of pH of the Drug Microenvironment
The ionization of a drug depends on the pH of the surrounding medium and the pKa value of the drug. Salt formation is not possible for unionized compounds; therefore, adjusting the pH of the drug microenvironment is an effective method for improving solubility of ionizable drugs.
This can be achieved in two ways:
Changing the microenvironmental pH alters the ionization behavior of drugs and is one of the simplest and most widely used techniques to increase aqueous solubility. According to the pH-partition hypothesis and Henderson–Hasselbalch equation, drugs remain in ionized form depending on gastrointestinal pH conditions, which improves dissolution and absorption.
In general:
Eutectic Mixtures
In eutectic systems, two components are melted together to form an intimately mixed physical combination. The components show complete miscibility in the molten state but very limited solubility in the solid state.
Unlike solid solutions, eutectic mixtures consist of finely dispersed crystalline components that enhance dissolution due to reduced particle size and increased surface contact area between drug and carrier materials [22,23,24].
Use of Co-solvents
Co-solvent systems significantly increase the aqueous solubility of poorly soluble drugs. A co-solvent system consists of a mixture of water-miscible solvents used to dissolve lipophilic drugs.
Common biocompatible co-solvents include:
Etman et al. [25] studied the solubility enhancement of etodolac using different co-solvents such as ethanol, propylene glycol, PEG-400, and glycerol. They also evaluated sugars (sucrose, sorbitol, mannitol), hydrotropic agents (sodium benzoate and sodium salicylate), and surfactants (Tween 80 and Brij 58).
Based on solubility data, a preliminary formulation containing 100 mg drug in 3 mL aqueous co-solvent mixture was developed for parenteral administration. The formulation was evaluated for clarity, turbidity, and precipitation behavior.
Self-Emulsification Drug Delivery System (SEDDS)
Self-emulsification is a technique in which a mixture of oil, surfactant, co-surfactant, and one or more hydrophilic solvents forms a clear isotropic system without the need for an external aqueous phase. This system is known as a Self-Emulsifying Drug Delivery System (SEDDS). When the formulation comes into contact with aqueous media such as gastrointestinal fluids, it spontaneously forms an oil-in-water (O/W) emulsion or microemulsion. This process improves the dissolution and absorption of lipophilic drugs.
The self-emulsification process depends on several formulation factors, including:
Self-emulsification occurs due to easy penetration of water into liquid crystalline or gel phases formed at the surface of droplets [26].
Several parameters are used to evaluate self-emulsifying performance, such as:
Among these, droplet size plays a crucial role because it determines the rate and extent of drug release and absorption. Positively charged emulsion droplets can be produced by adding small amounts of cationic lipids. Studies have shown that positively charged emulsions significantly improved the oral bioavailability of progesterone in animal models compared with negatively charged formulations.
Advantages of SEDDS
Limitations of SEDDS
Liquisolid Technique
In the liquisolid technique, poorly soluble or water-insoluble drugs are dissolved or dispersed in a non-volatile solvent and then converted into free-flowing powders using carrier and coating materials, as proposed by Spireas and co-workers. Solubility is a key factor for achieving the desired drug concentration in systemic circulation and obtaining an effective pharmacological response.
Liquisolid compacts are compressible powder forms of liquid medications. One of the major challenges in modern pharmaceutical research is improving dissolution, absorption, and bioavailability of poorly water-soluble drugs [29].
Approaches for Improving Drug Solubility
Liquisolid systems contain liquid medications such as oily drugs or suspensions of poorly soluble drugs dissolved in suitable non-volatile solvents known as liquid vehicles.
The therapeutic effectiveness of a drug mainly depends on its bioavailability, which is directly related to drug solubility and dissolution rate. These parameters are essential for achieving the required drug concentration in systemic circulation [30,31].
Melt-Granulation Technique
In the melt-granulation technique, powder drug particles are agglomerated using a molten binder. The binder may be a molten liquid or a solid material that melts during processing. This process is commonly carried out in high-shear mixers, where the temperature rises above the melting point of the binder either through external heating or mechanical energy generated by the impeller.
A major advantage of melt granulation is that it does not require water or organic solvents, and no drying step is needed. Therefore, the process is environmentally friendly and reduces manufacturing time.
CONCLUSION
In conclusion, solubility plays a critical role in drug formulation and development. The oral absorption of poorly water-soluble drugs is mainly limited by their dissolution rate. Therefore, improving drug solubility is an essential requirement for designing effective dosage forms.
Various techniques discussed in this review can be used individually or in combination to enhance the solubility of poorly soluble drugs. Many drugs show reduced bioavailability due to solubility limitations, making solubility enhancement an important area of pharmaceutical research.
The selection of an appropriate solubility enhancement technique depends on drug properties such as chemical nature, physical characteristics, and pharmacokinetic behavior. With the advancement of modern pharmaceutical technologies, it is now possible to significantly improve the solubility and therapeutic performance of poorly soluble drugs.
REFERENCES


Jadhav Kshitija R.*, Milke Umed. R., Sheikh Sameer. S., Approaches for Solubility Enhancement in Pharmaceutical Drug Development: A Comprehensive Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 4, 624-632. https://doi.org/10.5281/zenodo.19411009
 10.5281/zenodo.19411009
10.5281/zenodo.19411009
