1Amity Institute of Pharmacy, Noida
2Sardar Bhagwan Singh University, Dehradun, Uttarakhand
A major contributor to the process of drug distribution in the body is the affinity of the compounds for plasma proteins and bodily tissues. Pharmacokinetics Systemic circulation is the absorption site of drugs in the body, mainly existing in the form of drugs-complex and free. There is a dynamic equilibrium between bound drugs and free drugs. Free or unbound drugs readily cross membrane barriers. The bound drugs are still used as depots and slowly released to free drug. One of the most important physicochemical properties that can influence pharmacokinetics and therapeutic efficacy is the extent to which drug bind to plasma proteins. So, the fraction bound to plasma protein is one of the main determinants of drug distribution. Human serum comprises approximately 60% of the total plasma proteins in the blood. Proteins such as Albume (HSA) can bind various distinct substrates and the latter potentially affecting the %PPB. Classic methods for determining PPB, such as equilibrium dialysis and ultra-filtration, typically operate on time scales of minutes to hours, ruling them out for determination of compounds that undergo significant chemical degradation over time. Moreover, such methods are tedious and cannot be automated. There have been several attempts to develop computational models for prediction of chemical binding to plasma proteins. The present work aimed at developing prediction models on available Artificial Intelligence (AI) based algorithms that can accurately predict the drug-plasma protein interaction. Here are a few aspects and trends that should come in the future as technology and research in this field advance: Deep learning neural network Graph neural network Multi-modal data integration Explainable (AI) Generative models Transfer learning Personalized Medicine Data augmentation and synthesis High-Performance computing Ethical and Regulatory considerations.
The therapeutic properties of a drug are influenced by how it interacts with the proteins of blood plasma. This means the drug’s pharmacokinetics and pharmacodynamics — its absorption, distribution, metabolism, excretion and clinical effect as a medicine. Check how drugs bind to proteins in plasma at early stage of drug designing. Another key factor to test is the dissolution and partitioning of compounds, and their metabolic properties (1). There are many substances in blood plasma that may bind with medications, but two predominant proteins — human blood albumin and ?1-acid glycoprotein — are present in relatively high concentrations and can inhibit a wide variety of medications. This is because they are dense enough to have an outsized impact on how the drugs are disposed of, and how they function. Immunoglobulins and lipidemic proteins may also contribute a minor component (2–3). There are different methods that are used for the assessment of drug-protein interaction. Methods used to study drug-protein binding could be roughly separated into separative and non-separative techniques. The first category relates to the dissociation of the non-attached ligands from the attached mentioned organisms, which processes are used to quantify the free agonist or the number of adhered20.
The following group of functions begins with the detection of a change of a physical and/or chemical property of a protein or ligand due to the binding (5). The genomics is used to search significant genes in this huge amount’s information (6) that can use also to find desired proteins for drugs development and discovery. Try looking up proteins with some protein structure modeling tool. Computational intelligence (AI) and the application of machine learning methods have led to a phenomenal change in the field of pharmaceutical research and development. This is because drug–protein interaction prediction, sometimes called drug–protein binding prediction, is one of the key components of developing medicines. In this domain, accurate predictions are essential to enhance the pharmaceutical candidates, reduce costs, and speed up the drug discovery process. Currently, there are several AI techniques to predict the drug-protein interaction. It also explores the various applications of these techniques and the need for inventions in this rapidly developing field (7)
2. Techniques for drug/protein binding
Designed separative and non-separative methodologies for the drug-protein interaction studies are used. The first category involves differentiating the free ligand from the bound species, and is used to directly estimate the concentration of either the bound or unbound drug (8). The other category relies on identifying a change in a physicochemical property of the protein or the ligand upon binding (9).
Calorimetric and/or spectroscopic procedures, as reported earlier (10), are the methods of choice to gain detailed information on binding processes in the later stages of drug design or discovery. SPR-derived kinetic data is likely to play a key part in describing a wide range of biological events. For example, what has been a subject of debate for decades, is whether it should include the kinetics of drug-protein binding in establishing a drug PK property. In most cases, though, it seems not mandatory, given that the time scale for equilibration of protein binding is far shorter than that from other pharmacokinetic models, such as drug distribution and excretion. Consequently, in most realistic situations, the hypothesis of an instantaneously binding equilibrium holds (11). It is important to take into account the stoichiometry of the system, because certain techniques (SPR, spectroscopic methods,) work well when they are used to study a binding model with a simple 1:1 stoichiometry but either do not work or require complex data analysis for models which have many equilibria (12).
3. Artificial intelligence application:
3.1 Lead Compound Identification: AI models can rapidly screen millions of compounds and prioritize those with the highest binding affinity, thereby reducing the time and resources required.
3.2 Target Identification: AI-based methods can identify novel drug targets by analyzing biological data and detecting proteins that may be associated with specific diseases.
3.3 Poly-pharmacology: AI can help predict how a drug interacts with multiple proteins, which can aid in designing multitarget drugs to treat complex diseases.
Example: Personalized Medicine: AI can help recommend drugs based on the genetic profile of an individual, allowing for more effective treatment and fewer side effects.
3.4 Drug Repurposing: AI discovers new applications for existing drugs by predicting how they will interact with different proteins. This might hasten the process of developing therapies for rare diseases. More information on Patents and Intellectual Property in AI for Drug-Protein Binding (13).
4. AI continues to thrive with its efficiency and accuracy.
AI faces significant data challenges, such as data scale, growth rate, variety, and latency. Some databases that study drugs in the field of medicine can catalog thousands upon thousands of chemicals. Such large datasets are difficult for conventional machine learning algorithms to process. Computer modeling based on quantitative computer-activity relationship (QSAR), allows for the rapid prediction of multiple compounds or fundamental physical properties, such as (logP or logD), but these theories still do not possess the ability to predict the complex biological properties of chemical molecules, such as the potential benefits and potential side effects. Additionally, it is important to mention that algorithms rooted in QSAR face specific hurdles such as the restricted collection of learning information, possible inaccuracies in tests resulting from the experiments used to create the algorithms, and the lack of laboratory confirmations. These challenges may be overcome with the application of modern artificial intelligence techniques like deep learning and appropriate modeling studies to evaluate the efficacy and effectiveness of the pharmaceutical agents. These assessments depend on the use of large datasets for programming and analytic functions. In late 2012, Merck sponsored a Deep Learning Machine Learning Challenge on quantitative structure-activity relationship capable of making predictions about the suitability of a compound for drug development in a medical manufacturer setting. The predicted results using deep learning methods show a more professional prediction accuracy than traditional machine learning methods, with a better prediction effect on 15 database data related to the absorption, distribution, metabolism and toxicity of drug candidates [14, 15], The simulated chemical environment is large, which can show a particle area, showing the distribution map of particles in space and their corresponding properties. This process of collecting spatial data of components within a chemical-like zone aimed toward cells that are bioactive, is represented to follow this below. The identification and selection of appropriate components for more detailed experimental testing is improved by utilizing simulated scanning. Examples of freely available biochemical databases, are “Pub Chemistry”, “ChemBank”, “DrugBank” and “ChemDB”. Simulated screening of compounds inside computational molecular spaces can be done using numerous structure- and ligand- based computational methods. This decreases the expense, yet improves profiling evaluation, quick exit of nonlead atoms and better choice of therapeutic atoms [16]. Multiple factors may be leveraged in predicting the intended molecular architecture of a drug, such as prediction designs, molecular similarities, the process of molecular synthesis, and the use of in situ methodologies (17, 18). Pereira et al. proposed a Lenghty “DeepVS” program for forty sites and three thousand agonists. It was highly effective in the evaluation of the people, the system led to assess deception for ninety-five thousand people giving the above-mentioned sites [19]
5. Target identification
5.1 Prediction of the physicochemical properties by AI:
The way a drug is made physically—its dissolution, partition rate, amount of ionization, and internal permeation—affects how it works with certain target families without the drug's makers intending it to. Therefore, these characteristics should be taken into account throughout the process of creating a novel medication (20). Various artificial intelligence technologies may be utilized for the prediction of physical and chemical features. For instance, machine learning utilizes extensive database sets generated by prior complex refinement processes for training the algorithm [21]. Several chemical descriptions are used in drug design technologies. These include Smileys characters, possible energy measures, the densities of electrons around the molecule, and the locations of elements in three dimensions. These chemical descriptors are employed in conjunction with deep neural networks to produce viable compounds and then forecast their attributes [22]. Artificial systems that use ADMET predictions and ALGOPS software [23] are being used to guess how much water different substances have and how they will dissolve. Deep learning techniques, which include unsupervised chart recurrent neuronal nets and vertex-based convective neuronal nets training, were utilized in the prediction of molecular dissolution [24]. In their research, Kumar et al. constructed six prediction designs for estimating intestine absorbance. The frameworks employed multiple computations, which include support vector machines, fabricated nets, k closest neighbour computations, linear discriminant analysis, unpredictable artificial networking methods, and incomplete smallest circular regression. These predictions were trained using a dataset of “746” substances and subsequently tested on a separate dataset of “497” substances. Several cellular factors were used to make the predictions. These included genetic dimension, cellular weight, overall hydrogen number, atomic refractivity, cellular amount; log P, overall charged surface area, the sum of E-states measures, dissolution score, and the number of bonds that can be moved around. This approach aimed to accurately estimate the intestine absorbance of the molecules depending on their chemical features (Kumar et al., year, p. xx). In the same way, researchers have built-in computational algorithms designed around RF and DNN techniques to ascertain the gastrointestinal uptake of various biochemical substances [25]. Artificial intelligence plays a crucial role in the advancement of pharmaceutical development and research.
5.2 Prediction of bioactivity by AI:
You can use online tools like “ChemMapper” with the resemblance aggregation method to guess how drugs will interact with their targets [26]. Several machine learning algorithms, as well as deep learning algorithms, are being utilized for calculating drug-target binding affinity. These systems include “KronRLS”, “Symbols”, “DeepDTA”, and “PADME” machine learning methodologies, including Kronecker-regularized lowest methods, which are used to assess the similarities among medicines and peptide components in order to ascertain drug-target binding affinity. In the same way, “SimBoost” uses logistic models as a means to forecast DTBA taking into account both feature-based algorithms and similarity-based associations. Medication characteristics may be assessed by using several methods, such as receptor maximal usual substrate, increased interconnectivity thumbprint, or an amalgamation of these approaches (27). Deep learning techniques have demonstrated enhanced effectiveness in comparison to machine learning techniques. This improvement may be attributed to the utilization of network-based strategies in DL, which do not rely on the presence of the three-dimensional peptide layout [28]. “DeepDTA”, “PADME”, “WideDTA”, and “DeepAffinity” constitute a few deep-learning methodologies utilized for the quantification of drug-target binding capacity. These are capable of processing medication information in the manner of a simplified molecular intake line entry system. In this format, the peptide pattern is provided as the record information for proteins, while the medication shape is represented in a one-dimensional format [29]. “WideDTA” is a deep learning method based on convolutional neural networks that use different types of intake information, such as the receptor-simplified molecular intake line-entry system, peptide chains, ligand-based molecular fingerprints, and peptide regions and designs, to figure out how well the binding works (30). The methodologies of PADME bear a resemblance to the aforementioned techniques [31]. It is a comprehensible model for deep learning that leverages a combination of recurrent neural networks and convolutional neural networks, as well as both untreated and marked input. The analysis incorporates the substance represented in the Simplified Molecular Input Line Entry System layout as well as the molecular structure and physical characteristics of peptide patterns [32]. PADME technology is a DL-based system that makes use of direct networks known as feed-forward to provide predictions on DTIs. The proposed methodology takes into account the amalgamation of the medicine's characteristics and the desired protein properties as initial information and predicts the magnitude of their association. The smile depiction and the amino acid component are utilized to depict the medication and the objective itself, respectively [33]. Unauthorized machine learning methods, including MANTRA and PREDICT, have the potential to anticipate the beneficial effects of medicinal products and identify desired amino acids for both existing and upcoming medications. These methods can also be used to help re-use medicines and figure out the biological processes that make their beneficial effects possible. The MANTRA algorithm uses a “cmap” database to put substances together according to their shared patterns of gene expression. The process then moves on to grouping substances that are thought to work in the same way and follow the same biochemical pathway, as explained in reference [30]. The assessment of a medicine's biological function consists of an examination of its absorption, distribution, metabolism, and excretion statistics. A number of AI-based methods, including “XenoSite”, and “FAME” are used to find the exact spots inside a drug where decomposition takes place. Additionally, many computer programs, including “CypRules”, “MetaSite”, “SMARTCyp”, and “WhichCyp”, have been used to find the exact copies of the cytochrome P450 enzyme that helps the metabolism of a medicine. It was possible to figure out the clearance route for “141” approved medications using SVM-based predictions, which were very accurate [34].
5.3 Prediction of the target protein structure:
In the process of therapeutic compound development, the accurate identification of the receptor is crucial in order to achieve favorable outcomes. The pathogenesis of the illness involves the participation of a multitude of amino acids, which in certain situations exhibit heightened expression levels. To reach specific goals for diseases, it is necessary to accurately predict the structure of the amino acid that is being targeted in order to make the therapeutic compound more easily. Through its ability to guess the three-dimensional form of amino acids, AI could help with the discovery of structurally dependent drugs. It is useful to be able to predict things because it lets you make compounds that fit with the chemical structure of the peptide of interest. Because of this, AI might help in predicting how an ingredient will affect the protein of interest and taking safety concerns into account before the substance is made [35] Using AlphaFold, an AI method based on deep neural networks, made it easier to look at how far apart proteins are in space and how the polypeptide connections that connect them are oriented. This analysis aimed to forecast the three-dimensional form of the desired protein, yielding commendable outcomes as it accurately predicted “25” out of “43” molecule configurations. AlQurashi used recurrent neural networks to conduct the prediction of amino acids. The writer examined a repetitive geometrical system that includes three distinct phases: computations, mathematics, and evaluation. In this study, the encoding of the essential amino acids came first, followed by the consideration of rotational degrees for a particular region. These positions were acquired through the geometrical component located proximal to the component under investigation. Subsequently, the collected structural orientations were used as feed to generate a fresh core structure as a product. The ultimate module generated the three-dimensional configuration as the resulting outcome. The evaluation of the disparity between anticipated and tested configurations was conducted utilizing the dRMSD tool. The settings of the RGN model were tuned in order to minimize the root-mean-square deviation among the experimentally determined configurations and the anticipated features [36]. AlQurashi hypothesized that his artificial intelligence methodology would exhibit superior efficiency compared to the computational energy required for peptide conformation prediction. Nevertheless, it is probable that Alpha Fold will exhibit enhanced precision in the prediction of peptide layouts that possess patterns closely similar to the original forms [37]. A research investigation was undertaken to forecast the two-dimensional configuration of an amino acid using MATLAB, facilitated by a unique three-layered neural network toolkit that depends on a feedback-controlled instruction approach and a replication-erroneous approach. Neural networks were used for machine learning and outcome assessment, and MATLAB was used to train both the incoming and final databases. The predictive precision for determining the two-dimensional framework was found to be approximately sixty-three percent [38].
6. Predicting drug-protein interactions:
Being able to accurately guess how a pharmaceutical compound will interact with a binding site or amino acids is a key part of understanding how well it works. This capability also facilitates the reuse of medications and serves as a preventive measure against polypharmacy (39). Several artificial intelligence techniques have been shown to be good at correctly guessing the links between ligands and proteins, which makes the therapy work better (39, 40). In their study, Wang et al. presented an algorithm that used the support vector machine technique. This algorithm was to identify and investigate the connection between nine fresh substances and four important domains (41). This research funding was described as follows: In their study, Yu et al. used two algorithms to forecast possible relationships between drugs and proteins. This was achieved by combining pharmacology and chemical-based information and then evaluating the prediction versus established channels. The RF systems shown had an elevated degree of sensitivity and specificity in their performance. Furthermore, these methods showed that they could predict connections between drugs and targets. These predictions could then be expanded to include connections between targets and diseases as well as correlations between targets. Consequently, this accelerated the process of creating medications (42). Xiao et al. used MAT in order to gather refined information for the following advancements in drug targets. The proposed model consists of four distinct sub-predictors that are designed to detect and anticipate relationships among drugs and various biological targets, namely receptors to G-protein channels of ions, enzymes, and nucleotide regions accordingly. Using target-jackknife tests to compare this generator's performance to that of other models that had already been made, showed better results in terms of accuracy and consistency.
Artificial intelligence has been used to predict the relationships between medicines and their targets. This has also been used to make it easier to reuse old drugs and reduce the incidence of poly-pharmacology (43). The reuse of a current medicine renders it eligible for immediate inclusion in the second stage of human studies [44]. Additionally, there is a reduction in expenses since the reintroduction of an already-available medicine incurs an expense of $8.4 million, but the introduction of an entirely novel medicinal item charges $41.3 million [45] The "remorse by connection" strategy may be employed to predict the potential relationship between a medicine and an illness. This method can be dependent on either knowledge or programmatically generated networks [46]. The utilization of machine learning methods, including support vector machines, neural networks, and deep learning, is prevalent in technologically controlled networks. Statistical regression techniques that make use of PREDICT and other machine learning algorithms look at a wide range of different things. Some of these things are how similar the molecules being studied are to each other, what those molecules are made of, and how gene expression changes when medications are recycled. [47]. The utilization of cell network-based deep learning technology has been investigated for the purpose of predicting the medicinal uses of topotecan, a topoisomerase antagonist that is now employed in medical practice. Furthermore, the inhibition of human retinoic acids has been identified as a potential therapeutic approach for the treatment of multiple sclerosis-related disorders [48]. The present system is now protected by an interim copyright granted in the United States. Autonomous models belong to the uncontrolled machine learning group and find application in the field of medication recycling. A ligand-based technique is implemented to identify new unwanted targets for a certain group of pharmacological components. This strategy involves conditioning the algorithm using a predetermined amount of substances that possess known physiological functions. The trained method is then utilized to analyze additional substances [49]. In current research, deep neural networks were utilized to reuse pre-existing medications that have shown efficacy against the SARS coronavirus, the HIV virus, and the virus that causes influenza, as well as pharmaceuticals that function as enzyme-inhibiting medicines. The artificial intelligence technology was trained using expanded connection fingerprinting along with an octanol-water partition factor. On the basis of the findings, it was determined that thirteen of the medicines that were examined showed potential for future improvement, as evidenced by their observed cell death and transmissible inhibitory properties (50).
When a pharmacological component binds to many binding sites in the body, it can cause unwanted and harmful effects [69]. Understanding the connections between pharmacological compounds and proteins can help predict the occurrence of poly-pharmacology. Using the principles of poly-pharmacology, artificial intelligence (AI) can make new compounds, which helps in the development of drugs with better safety profiles [70]. When combined with large datasets, artificial intelligence (AI) tools like self-organizing maps can help find links between a few chemicals and a lot of different chemical targets and off-targets. Bayesian classification and computations have been shown to be a good way to find links between the pharmacological properties of drugs and the possible receptors they bind to [49]. Li et al. (year) presented a study showcasing the utilization of “KinomeX”, an artificial intelligence (AI)-a powered web page that utilizes deep neural networks to recognize the poly-pharmacology of mediators by analyzing their molecular arrangements. The present technology employs a deep neural network that has been trained using a dataset consisting of fourteen thousand biological function observations. This dataset has been specifically constructed according to the analysis of over three hundred kinases. Therefore, this technique has significance in the examination of a medicine's sensitivity for the protein class as a whole, as well as specific subdivisions of kinases. Consequently, it aids in the development of innovative chemical moderators. In this work, it was utilized as a representative chemical to accurately anticipate its principal target as well as its missed targets [51]. An example worth mentioning is the cloud-based AI device that employs proteome-screening techniques. This tool is utilized to identify sensors capable of interacting with a certain short molecule, as described by its Smiles strings, and to assess both intended and unintended associations. This facilitates comprehension of the potential negative consequences associated with the medication [52]. The use of artificial intelligence (AI) in the process of completely novel drug design in the past few decades, a completely new pharmaceutical creation technique has gained significant popularity as a means of designing novel therapeutic compounds. Due to the inherent drawbacks of the previous approach, such as complex synthesis pathways and difficulties in accurately predicting the biological function of newly synthesized molecules, emerging deep learning (DL) technologies have replaced it [39]. Computer-aided synthesis preparation has the capability to propose a vast number of compounds that can be produced, as well as anticipate many biosynthesis pathways for these molecules [53]. The software called Synthia, was created by Grzybowski et al. [54]. The software is capable of encoding an array of principles into an algorithm and suggesting potential methods for creating eight medically important sites. The strategy has shown efficacy in enhancing crop productivity and minimizing costs. Additionally, this technology has the ability to provide alternative creation approaches for trademarked items and is purportedly advantageous in the creation of molecules that have not previously undergone production. In the same way, deep neural networks are primarily concerned with the principles governing organic chemical synthesis. By using representational artificial intelligence techniques, it facilitates response detection and medicinal product development and development procedures, surpassing the efficiency of conventional methodologies and expediting [55, 56]. A theoretical framework by Coley et al. suggested using a fixed anticipatory response pattern to create a group of reacting substances that can make physically useful goods at a fast rate of response. By using the ranking given by neural networks machine learning was used to find the most popular item. Putin and colleagues conducted a study on a deep neural network framework known as renewed adversary network computation. The purpose of their research was to investigate the application of reinforcement learning (RL) in the completely novel development of tiny chemical compounds. The training of this system involved the utilization of compounds that appeared in the shape of SMILES sequences. Subsequently, the system produced compounds possessing predetermined molecular descriptions, specifically the molecular weight, partition coefficient, and topological surface area of polar molecules. The performance of RANC was evaluated in comparison with a different system, organics. The results indicated that it had superior performance in developing distinctive designs while minimizing any significant reduction in structural dimension [57]. The use of the Long-term Memory model, which was based on “CHEMBL” library molecular information and displayed as strings, had an impact on the growth of the Recurrent Neural Network. The aforementioned approach was employed to create a comprehensive collection of compounds for virtual screening purposes. This methodology was expanded to acquire new compounds with a specific focus, which includes drugs for the serotonergic receptor in question, the bacteria Staphylococcus, and Protozoa falciparum [58]. Popova et al. developed a stronger understanding of structured evolutionary techniques for completely novel pharmaceutical manufacturing. This approach utilizes procedural and forecasting deep neural networks to facilitate the creation of novel molecules. The generation utilized in this study demonstrates a higher capacity for generating a larger number of distinct compounds, by using stacking storage. Conversely, anticipatory techniques are utilized to anticipate the characteristics of the synthesized substance [59]. Merk et al. used a creative artificial intelligence framework to create retinol and peroxisome proliferator-activated receptor ligand compounds that showed good treatment benefits without the need for complicated rule-based methods. The researchers were able to come up with five effective compounds, and four of them had strong modulating effects in cell tests. This shows how useful artificial intelligence can be for coming up with new compounds [60]. The integration of artificial intelligence in the process of creating a completely novel molecular layout holds potential benefits for the drug industry. This is because it has many benefits, such as making understanding easier, improving what you already know, and suggesting ways to make substances. As a result, artificial intelligence can expedite the process of lead design and growth in a more efficient manner [58, 61].
7. AI Technique for drug-protein binding prediction:
The primary objective of machine learning and artificial intelligence technologies is to uncover latent content and insight inside datasets. In order to enable machine learning algorithms to acquire the ability to execute various assignments, the technique is provided with trained information. The simulation has the capability to perceive and acquire information gathered from the retraining facts in an autonomous manner. The machine learning algorithm can find hidden patterns and connections in data that might not be obvious to human observers, especially domain experts. The verification of a database is a distinct piece of information frequently utilized throughout the learning process to evaluate the performance of the model and fine-tune its hyperparameters. Once the algorithm has undergone comprehensive development and validation, it is possible to evaluate its function by applying evaluation measurements to a self-sufficient sample database that was never utilized during the course of learning. This enables the monitoring of the system's predictive capabilities. Consequently, the acquisition and use of knowledge play a crucial role in the context of machine learning technologies. Overall, the efficiency of a machine-learning algorithm in terms of understanding and anticipating tends to improve as additional information is made available to it. The databases frequently utilized in this particular field include CASP targets, “LigAsite”and “PDBbind”. It's important to note that a number of statistics have been made using the Peptide Database Library's main amino acid form and receptor information [62]. In general, these collections are very useful as tools for people who want to test and improve machine learning-based algorithms that predict how proteins and ligands will interact, as well as for people who want to find new drugs.
8. Ligand Attachment Sites: Advanced Insights and Techniques
The receptor connection location is a list of easy-to-find interaction points in peptide shapes that are important for medicine. An approach is utilized to effectively eliminate physiologically unnecessary agonists by taking into account the agonist's quantity of heavier molecules and the amount of interparticle interactions between the peptide and agonist [63]. The presence of two loose and attached configurations for every peptide in the database confirms its suitability for evaluating the performance of interaction region prediction algorithms. Edition “9.7” includes a compilation of approximately “390” amino acids, whereas the repetitive table provides details about a total of eight hundred and sixteen molecules.
Several studies have been done to examine the binding site prediction techniques, binding affinity, and available ligand-binding site due to the availability of various effective Al algorithms and the amount of dataset in this area. Figure (2) depicts a simple instance of the ML approach in protein-ligand interaction prediction.
The characteristics of the target protein and ligand are determined first, followed by data preparation stages. Normalization (a data processing method) is widely employed in machine learning because it transforms the numeric columns values in a data set to a comparable range without altering the scales of results or missing data. Normalized dataset is then supplied into a machine learning algorithm, including the neural network displayed in the figure. This phase is frequently taken as empirical evidence shows that data normalization enhances accuracy [64] Multiple procedures operate at hidden levels, and the expected decision is finally output by the output layer. There has been a great deal of interest in utilizing ML and especially potent deep learning approaches to predict PLIs since it does not require specific hard-coded rules developed by human specialists to make predictions and may provide excellent accuracy in prediction.
This library is a meticulously maintained collection of drug-protein interaction events that have significant pharmacological relevance. After the automatic process of figuring out how scientifically important a binding agent is, a full qualitative analysis is done to fix any mistakes that might have happened. The primary source of structural information is derived through the Protein Dataset, while additional information related to biology is acquired from academic research and various additional sources. A careful verification process is conducted to identify potential misleading results. This involves thoroughly examining the initial research and cross-referencing it against additional resources. This rigorous procedure guarantees the integrity and excellence of the dataset [65]. The database undergoes periodic updates, with the latest edition including an overall total of fifty thousand elements. These records consist of many categories, including ten thousand peptides from the amino acid Data Bank, fifty-seven thousand Deoxyribonucleic acid agonists, twenty-six thousand amino acid agonists, ten thousand metallic agonists, and thirty thousand ordinary molecules. Among the available records, a total of twenty-four thousand possess attachment-sensitive knowledge.
The Bound “MOAD” file, which is often called the "root" of the whole database, is a complete set of great peptide-ligand relationships gathered from the Peptide Information Network. Information gathered from scientific publications has expanded this informational collection. The choice of Bound MOAD, a database on molecular interactions, is to prioritize compatibility information based on the dissociation constant above the inhibition constant (Ki) and the half-maximal inhibitory concentration (66–67). The layout of the system used a 'well-secured' strategy to ensure the inclusion of all peptide-drug complexes possessing a three-dimensional configuration. Periodic revisions are conducted on a yearly basis with the purpose of incorporating additional interactive sensitized data that becomes accessible in the Protein Information Bank. The present iteration of the dataset has a total of thirty-eight thousand receptor-ligand building components, accompanied by fourteen thousand interaction records. Additionally, the dataset comprises a thousand distinct agonists and encompasses a diverse range of ten thousand polypeptide categories.
Grading systems are often used to figure out how protein-ligand interactions work in the context of substance layout based on structure. Numerous grading systems were developed extensively, necessitating the need for public availability benchmarking to evaluate their efficacy and limitations. The CASF benchmarking gives an idea of what the expected results will be from known grading operations ahead of time. This allows for an accurate evaluation of the algorithm compared to current evaluation methods that use the same set of experiments. The outcomes of the evaluations included relying on a curated collection of 284 peptide-drug interactions that possess excellent crystallized arrangements and dependable interaction statistics [68]. The sample dataset used in this study was derived from “PDBbind” improved collection, namely edition 2016. The assessment techniques in CASF-2016 have experienced enhancements as compared to their predecessors. The outcome assessment of a grading mechanism is conducted using four indicators: grading strength, ranked authority, docked strength, and testing potential.
9. Prediction of ligand binding site of protein
In the bigger picture of finding new agonists, simulated evaluation needs to know exactly where on the target the drug binds. However, in certain instances, this essential data remains unidentified. Finding the precise locations on amino acids where enzymes and ligands attach requires looking at their three-dimensional arrangement. This is very important in the field of structure-driven chemical element development (69, 70). This method also contributes to the anticipation of medication adverse reactions (70) and enhances our comprehension of a molecule's functionality (71). The specific placement of peptide remains inside the molecule, which is usually found in small areas, shows how peptides and agonists are connected. It is important to find these important areas in order to understand how proteins work, learn more about the relationships between cells, and make loading simulations easier for drug discovery based on simulations. The peptide remains in structures that possess particular significance, often referred to as the Ligand Binding Sites. Experimental investigation has shown that the interaction region for the agonists of an amino acid is often located inside the largest area on its outermost layer (72). A study using an experimental set of peptide layouts discovered that the SURF-NET design [73] was the best at figuring out 83% of the time where the drug would attach. The results obtained indicated that the interacting domain was located inside the biggest gap of all ten peptides that were examined. The outcome obtained was identical.
Every individual peptide, also known as a remnant, has a unique influence on the overall form and functionality of a molecule. Despite a significant separation in the calculated separation among both sites in an amino acid series, it is possible for their physical proximity to be relatively close as a result of peptide packing [74–77]. Consequently, regions located away from the intended protein in terms of distance but in close proximity physically may have a substantial influence on the positioning of interacting sites. A fold is widely regarded as a significant advancement in the field of peptide framework prediction. It successfully integrates several protein properties, including one dimension, two dimensions, and three-dimensional knowledge, to reliably estimate the secondary geometries of a wide range of molecules. In the context of predicting attachment sites, it is important to take into account leftovers that are in close proximity in space. Moreover, the influence of additional structures of the peptide on interaction is frequently very important compared to that of its main shape.
10. Binding site prediction methods
Throughout the past twenty years, a variety of methodologies have been developed for the prediction of attachment sites. These methodologies include numerous methods, including the use of blueprints, power operations, mathematical principles, and machine learning techniques .The template-based methods attempt to predict the position of binding sites on an input protein using known protein templates. They are based on the assumption that proteins sharing a similar structure can also share a similar function [78]. In comparison to the geometry and energy-based methods, these methods are generally more accurate if a good template can be found [79].
This approach aims to forecast the location of interaction domains on a given peptide using existing peptide patterns. These findings are predicated on the underlying premise that molecules exhibiting a comparable structural arrangement are likely to possess identical functional characteristics [78]
For structure-based approaches, one way to find the planar cavity on the desired amino acids is to look at the layout of the cellular interface [79]. A group of people divided the method for finding reservoirs into three groups: matrix imaging, probing globe-stuffed, and alpha structure.. Layout-based approaches include the analysis of the chemical interface's structure in order to identify exterior voids present on the desired molecule. Two main types of techniques used in chamber labeling are matrix imaging and sensor spherical packing. The other two are in alpha form. The matrix-based imaging technique involves enclosing the amino acid inside a 3D matrix. Subsequently, spots within the framework are identified as pockets if they satisfy specific geometrical criteria. The precision of this approach relies on the clarity of the matrix. Sensor-based spherical techniques include the literal application of specialized sensor circles to stuff voids. In addition, alpha-shape methodologies depend on the calculation of triangles [80] in order to identify empty spaces on the outermost layer of the peptide. The ultimate stage of each of the above methodologies is the process of grouping and a subsequent rating mechanism for various areas that have been detected.
11. Network-based methods
We categorized network-based methods, which utilize graph-based methods to perform the DPPI prediction task (Figure 3). The simplest highest accurate inference methods based on network, network-based inference (NBI) for DPPI prediction uses only DT bipartite networks topology comparability [81]. Additionally, a variety of strategy integrates three networks of drug-drug similarity, protein-protein similarity and existing DPPI into a heterogeneous network, and predicted similar drugs often target similar proteins [82]. Aside from conducting the DPPI prediction task, a two-layer unstructured graphical representation of the network has the potential to be utilized to train on simply the prediction of direct DPPI (typically caused by the binding of a ligand to a protein), passive DPPIs, and mode of action of the drug (binding interactions, activating interactions, inhibiting interactions). Another example is given in [83] using the Restricted Boltzmann Machine (RBM) [84].
For prediction, these traditional pioneering binding prediction methods use existing templates and/or genetic identity or molecular geometry to predict the binding pockets. COACH and its improved Edition D exploited the PDB by following template edge predictions outputted from previously solved structures of the target complex [85, 86]. Another widely used server, “LIGSITE” uses geometry by scanning the model within a 3D grid map for convolutions by defining grid points in order to define if the location is a solvent of the protein eventually identifying possible binding sites [87]. Of course, there are limitations to the use of these concepts. The predictions become less accurate when the pair wise identity of the target to the template reduces and generally becomes unreliable when the identity approaches the ‘twilight zone’ [88, 89]. Template-based methods are quite restrictive for novel protein folds. As the field advanced, Find Site was one approach that used structural similarity to find templates. From biology point of view, the theory of utilizing structure and sequence similar proteins as prediction templates rests on few grounds such as proteins undergo mutations over years of evolution, therefore functional region tends to be more conserved even among the same family proteins as such mutations carry a risk of losing function [86, 89 and 90]. So care should be taken obtaining this way since it's not perfect conservation and there can be a lot of variability between the residues of the family or the function of very well conserved areas of the protein. [89]
So, template-based methods can succeed if a template with known binding site information is available but fail otherwise. Machine learning approaches are able to learn from existing data and generalize on new data that are not similar to the training data. Energy function-based approaches were probably trained on a small number of known protein-ligand structures with the function prepared by human experts, which may not be the best fit for a large amount of data and do not generalize well to new information. In the same vein, geometry-based methods identify surface cavities on a target protein by examining the molecular surface geometry, the exactitude of which is closely proportional to the grid resolution. (37).
Blueprint-dependent approaches succeed when they have blueprints with solid interaction websites, but they cannot succeed when they don't have the right ones. Techniques of computer intelligence have the capability of learning knowledge from the past data and using it when the novel data instances appear that might be far beyond the similar context of data used in the learning process. It is done only for very few well-known peptide-drug structures and is based on high standards from biological experts for the evaluation of power value-based methods. But this method may not generalize well to large amounts of information, and it may not be able to generalize well to new facts. Simultaneously, geometrical methods track membrane pockets in the material based on the geometric properties of the cellular interface. The depth of the matrix employed has a tremendous impact on the precision of these methods. However, this structure-dependent method is also prone to errors, especially when the rules from analysis are different or if the peptides in the matrix structure are not presented in the same way as with 91. Depth-based training techniques can fit a parametric function by using large sets of input consisting of many pieces of knowledge. They yield even more accurate predictions of how proteins will interact with one another.
A diverse set of datasets exists for quality peptide layouts. That has led to new forms of application using artificial intelligence techniques. The essential idea behind existing procedures of AI predicting where connections will happen could be sorted into five essential stages: gathering, securing, and readiness; highlight plan; organize arrangement; approval; assessment; and parameter tuningassessment. First, a comprehensive collection of known enzyme-ligand interaction datasets is gathered together into a single repository, and several important features will be extracted to represent the enzyme and receptor molecules. These representations are then normalized. Some artificial learning prototypes study and handle feedback skills to predict attaching locations. These mathematical approaches involve superficial guided mastering calculations, synthetic neurological systems, convex brain networks, and collective techniques, among others. Further details regarding these unique approaches are available under sections— Standard techniques for attaching location anticipating and In-depth creating techniques for attaching location estimation Machine-developing techniques may be specialized into two basic pair groups: traditional machine-acquiring approaches, which do not utilize calculations for techniques, and current profound experience procedures.
Table 1 summarizes commercially available software for prediction of the PPB of compounds. It is obtained through an in-depth internet search and extensive literacy review.
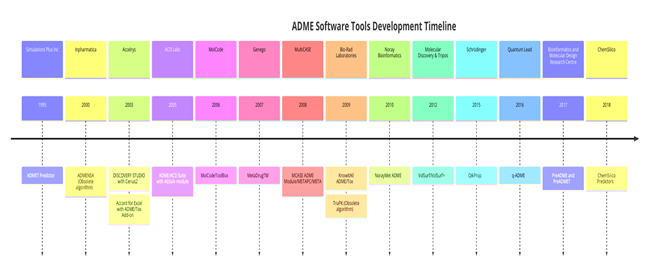
Figure 14.1: Commercially available software for prediction of PPB.
12. Recent Advances in AI for Drug-Protein Binding Prediction
In modern times, artificial intelligence has become known as a potent instrument for forecasting the outcome of molecule-peptide interactions. The development of algorithmic design, specifically convoluted brain nets and recurring brain nets, is a big step forward in the field. These models can look at a lot of data, such as peptide shapes and receptor attributes. The use of such methods has significantly enhanced the precision of bonding affinity estimations. In addition, the incorporation of atomic dynamical computations in conjunction with artificial intelligence has bolstered our comprehension of the kinetic elements associated with the connections between proteins and drugs. The use of artificial intelligence in simulated evaluation techniques has significantly expedited the process of identifying prospective medications. This is achieved through the effective examination of extensive chemical archives.
13. RECENT PATENTS
According to studies, artificial intelligence applications are becoming more prevalent in drug-protein binding prediction. These methodologies are meeting some of the organizations' goals, with notable gains in drug discovery, and among other areas. However, it remains to be seen if these techniques will be useful in assisting researchers in developing and identifying improved drug candidates more quickly (4, 18, 80).
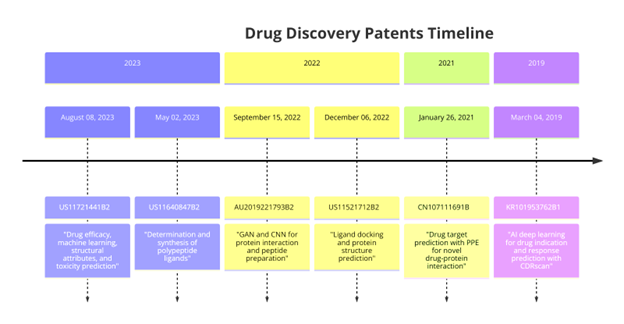
Figure 16.1: Issued AI patents for drug-protein binding prediction.
14. Conclusion
The interactions between drugs and receptors or proteins are essential for the pharmacokinetics and pharmacodynamics of therapeutics, affecting how they work, how they travel within the body, how they are digested, and how they are excreted. Methods to study such interactions are well-established in experimental research, such as equilibrium dialysis and ultra-filtration, which, although valuable, are labor-intensive, time-consuming, and not well-suited to handle chemically unstable products. With the introduction of artificial intelligence (AI) and machine learning, this field has witnessed a transformative change through the innovative, efficient, and precise methodologies for predicting drug-protein binding properties. Deep learning and neural networks are part of AI-based algorithms which have shown great promise in the modeling of complex biological interactions, binding affinities prediction and novel drug target discovery. Additionally, the utilization of AI techniques like generative adversarial networks, deep reinforcement learning, and convolutional neural networks has notably sped up the drug discovery process through virtual screening, lead optimization, and personalized medicine. Furthermore, such innovations decrease the cost and time of preclinical testing as well as improve the accuracy of predicting pharmacological effects. Although they hold great promise for transforming the field, data quality and diversity, interpretability, and ethical issues are some of the challenges that remain to be addressed. The convergence of AI technologies with high-performance computing, multi-modal data, and systems biology holds great potential to unravel drug-protein dynamics to develop precision medicine for pathology of drug-target interactions. AI-driven techniques, which are also constantly evolving, will inevitably make pharmaceutical research and development more dynamic, efficient, and patient-centric. Such advances in healthcare depend on collaboration between computational and experimental scientists, and must be supported by appropriate regulatory frameworks to reach their full potential.
REFERENCES


Vishal Rastogi, Ravi Maurya, Prakshal Jain, Advancing AI-Driven Approaches for Enhanced Prediction and Analysis of Drug-Protein Interactions, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 12, 2559-2578. https://doi.org/10.5281/zenodo.14534354
 10.5281/zenodo.14534354
10.5281/zenodo.14534354
