Department Of Pharmacology, Devaki Amma Memorial College of Pharmacy.
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cognitive decline, memory loss, and behavioral impairment. It is the leading cause of dementia, with a rising prevalence due to aging populations. The neuropathology of AD involves amyloid-beta (A?) aggregation, neuroinflammation, and cholinergic dysfunction, which contribute to neuronal damage and cognitive deficits. Despite significant research, there is no cure for AD, and current treatments provide limited benefits. In vitro models, including brain tissue cultures, stem cell-derived models, and molecular simulations, play a vital role in understanding AD pathology and testing potential therapies. These models help investigate key mechanisms such as A? accumulation, tau hyperphosphorylation, and neuroinflammation, providing insights into targeted therapeutic strategies. As AD continues to impose a substantial global health burden, ongoing research and innovative treatments are necessary to develop more effective therapies and improve patient care.
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cognitive decline, memory loss, and impairment in thinking and behavior. It is the most common cause of dementia, a condition that significantly interferes with daily activities and quality of life. AD develops gradually, worsening over time and ultimately leading to severe cognitive dysfunction [1]. The history of AD dates back to the early 20th century when Dr. Alois Alzheimer, a German psychiatrist and neuropathologist, first identified the disease. In 1906, he documented the case of Auguste Deter, a 51-year-old woman who exhibited severe memory loss, language difficulties, and behavioral changes. After her death, Dr. Alzheimer examined her brain and discovered abnormal protein deposits and nerve cell degeneration—now recognized as key pathological features of AD. The discovery of beta-amyloid plaques and neurofibrillary tangles provided critical insights into the cellular mechanisms driving neuronal degeneration. Further studies have revealed a complex interplay of genetic, environmental, and age-related factors influencing AD progression [1][2]. Today, AD is a significant global health challenge, with an aging population contributing to its growing prevalence. Early detection, improved care strategies, and innovative treatments remain crucial in addressing its increasing burden on individuals, families, and healthcare systems. The prevalence of AD varies across regions but is consistently higher among older adults. According to the World Health Organization (WHO), approximately 50 million people worldwide were living with dementia in 2020, with AD being the most common cause. By 2050, the number of dementia cases is expected to triple, exceeding 150 million. Regional differences in AD prevalence reflect variations in demographic trends, genetics, socioeconomic factors, and healthcare access, with developed nations experiencing higher rates due to aging populations and better diagnostic resources [3]. AD remains a challenging disorder with no cure, and neuroinflammation plays a critical role in its onset and progression. As a result, treatments targeting neuroinflammation are an area of growing research interest. Natural compounds, which tend to have fewer side effects than synthetic drugs, have attracted significant attention as potential therapeutic options[4]. At the neurological level, AD is characterized by the progressive loss of neuronal synapses and pyramidal neurons, with the hippocampus and neocortex—regions responsible for complex cognitive functions—being the most affected. Gender is also considered a risk factor for AD, as women account for two-thirds of all cases. This is largely attributed to their longer life expectancy, increasing their vulnerability to age-related diseases. However, the precise relationship between sex and AD risk remains uncertain, as epidemiological studies have yielded conflicting results. Some research suggests that sex hormones may contribute to AD susceptibility [5]. Factors such as hormonal changes in aging women, sexual dimorphism in brain development, gene-by-sex interactions, and differences in immune responses have all been associated with an increased risk of AD in women.
2. Neuropathology of Alzheimer’s Disease
The neuropathology of Alzheimer’s disease (AD) is highly complex and not yet fully understood. Several hypotheses attempt to explain its pathological mechanisms.
2.1. Cholinergic Dysfunction in the Central Nervous System
Cholinergic decline, marked by reduced acetylcholine (ACh) levels, contributes to cognitive deficits in Alzheimer’s disease (AD) by impairing synaptic transmission and promoting neuroinflammation.[7] Amyloid-beta (Aβ) worsens this dysfunction by inhibiting choline uptake and ACh synthesis. Whether ACh reduction is a cause or consequence of AD remains unclear, but understanding this could lead to better therapeutic strategies.[8][9]
2.2. The Amyloid Cascade Hypothesis
2.2.1. Amyloid Precursor Protein (APP) Processing
Amyloid precursor protein (APP) supports brain health, but its processing determines its impact. α- and γ-secretase produce neuroprotective peptides, while β- and γ-secretase generate toxic amyloid-beta (Aβ), linked to Alzheimer’s disease (AD).[10] Disruptions in this balance contribute to Aβ accumulation, making APP processing a key focus in AD research.[11]
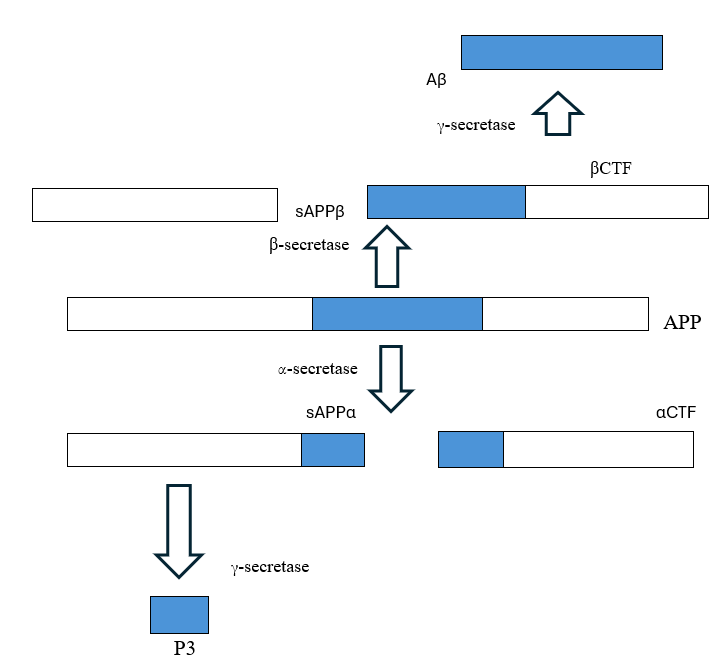
2.2.2. Structure of Aβ
Amyloid-beta (Aβ) aggregation drives Alzheimer’s disease (AD) pathology. An increased Aβ42/Aβ40 ratio promotes toxic fibril and plaque formation due to Aβ42’s hydrophobic nature.[12] Its shift to a β-sheet structure is key to neuronal damage, making Aβ aggregation a target for AD therapies[13].
2.2.3. Function of Aβ
Amyloid-beta (Aβ) has dual effects on neurons[14]. Low levels support memory, while excessive Aβ causes toxicity, leading to synaptic dysfunction and neuronal loss in Alzheimer’s disease (AD).[15] Balancing Aβ levels is key to developing effective AD therapies.[16]
2.2.4. Mechanism of Aβ Neurotoxicity
Calcium (Ca²?) balance is crucial for neuronal function, and amyloid-beta (Aβ) disrupts calcium channels in Alzheimer’s disease (AD). This leads to excessive Ca²? influx, mitochondrial dysfunction, and increased toxicity, contributing to neuronal damage. Understanding these mechanisms could guide neuroprotective strategies in AD. [17]
2.3. Neuroinflammation in AD
Neuroinflammation, triggered by Aβ deposition, involves chronic activation of microglia and astrocytes, leading to the release of pro-inflammatory cytokines, ROS, and neurotoxic molecules. This response contributes to neuronal damage and can further promote Aβ production, creating a self-perpetuating cycle of inflammation and neurodegeneration [18].
2.4. Other Pathogenetic Factors
Several factors increase the risk of Alzheimer’s disease (AD). The ApoE4 isoform of apolipoprotein E (ApoE) promotes amyloid deposition, oxidative stress, and neuroinflammation, contributing to AD progression.[19] Hypercholesterolemia is also linked to AD, with mutations in APP, PSEN1, and PSEN2 further exacerbating cholesterol-related pathology. Environmental factors may also play a role, though their impact remains under investigation.[20]
3. Drugs used to improve cholinergic function
Several drugs are approved to manage Alzheimer’s disease (AD), though their effectiveness is limited. Since AD follows a distinct pathological progression, stage-specific interventions may help slow its advancement. Some medications aim to enhance cholinergic function in patients [21]
3.1 Acetylcholinesterase Inhibitors (ACHEIS)
Acetylcholinesterase inhibitors (ACHEIS) were the first FDA-approved drugs for Alzheimer’s disease (AD), enhancing cholinergic transmission by preventing acetylcholine (ACh) breakdown. Key AChEIs include:
While AChEIs improve synaptic function, their benefits are modest, and limitations include side effects and high costs. Further research is needed to optimize their role in AD treatment.
3.2. M1 Receptor Agonists
The M1 muscarinic receptors in the brain remain largely intact in Alzheimer’s disease (AD), making them a potential therapeutic target. M1 receptor agonists, like, Xanomeline, show promise in improving cognitive function, though they may cause gastrointestinal and cardiovascular side effects. Milameline is another M1 receptor agonist used in AD treatment[23].
4. Anti-Aβ Drugs
Amyloid-beta (Aβ) peptides play a central role in Alzheimer’s disease (AD) by causing neurotoxicity, synaptic dysfunction, and neuronal loss. Aβ deposits disrupt calcium homeostasis and contribute to neuronal degeneration, making anti-Aβ drugs a key focus in AD treatment [24]
4.1 Calcium Antagonists
Calcium imbalances impact neurotransmission in Alzheimer’s disease (AD).
4.2 Antioxidants
Antioxidants protect nerve cells from oxidative stress in Alzheimer’s disease (AD).
4.3 Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
NSAIDs may help prevent, but not treat, Alzheimer’s disease (AD) by reducing inflammation linked to plaque formation. Indomethacin, tenidap, aspirin, ibuprofen, and naproxen have been studied, though long-term use risks liver and kidney toxicity[27].
4.4 Hypolipidemic Drugs
Apolipoprotein E (ApoE) influences Aβ deposition and plaque formation. ApoE isomers may reduce plaque buildup, suggesting hypolipidemic drugs as a potential AD treatment.[28]
4.5 Iron Chelators
Excess brain iron contributes to Alzheimer’s disease (AD) by generating free radicals and causing neuronal damage. Desferrioxamine, an iron chelator, helps prevent neurotoxicity but requires frequent dosing and may cause side effects like retinal toxicity.[29]
5. Vaccines for AD
Immunotherapy aims to prevent Aβ aggregation in Alzheimer’s disease (AD). AN-1792, the first AD vaccine, was halted due to autoimmune side effects. Passive immunization trials with anti-Aβ antibodies continue, but challenges remain.
Preventive, cost-effective, antigen-specific, widely effective, and ensure patient compliance.[30]
6. In vitro models of AD
In vitro models of Alzheimer’s disease (AD) offer a direct and efficient way to study AD pathology and test potential treatments at the molecular and cellular levels.[31]
Tissue model
Brain tissue cultures, such as hippocampal neurons, provide a platform to examine the effects of amyloid-beta (Aβ) peptides. A study by Yankner et al. found that Aβ fragments were neurotrophic to undifferentiated neurons but neurotoxic to mature ones[32]. These models also replicate AD-related changes, including inflammation, enzyme activity, and disrupted cellular processes, while offering controlled environments for testing therapies.[33] Metabolically active brain slices, such as those used in studies by Gong et al. and Li et al., enable the examination of AD-related tau hyperphosphorylation and neurodegeneration.[34] These slices better replicate the brain’s natural environment, offering a more accurate model compared to cultured cells. This approach allows for the study of AD at both biochemical and histological levels, as well as the testing of potential treatments like memantine.[35]
Cell models
Stem cell advancements have enabled the creation of disease-specific induced pluripotent stem cell (iPSC) lines from patients with familial (fAD) or sporadic Alzheimer’s disease (sAD).[36] These iPSCs provide a powerful model for studying AD and testing therapies. For example, iPSCs from fAD patients with mutations in PS1 and PS2 showed increased Aβ42 levels, supporting Aβ’s role in AD. Research also found that neurons derived from iPSCs of sAD patients exhibited similar phenotypes to those with fAD, highlighting the genetic complexity of sAD.[37] Treatment with docosahexaenoic acid (DHA) reduced Aβ levels in some cases, illustrating the potential for personalized treatments. Human neuroblastoma cell lines are also used to model AD pathophysiology. Studies have shown that these cells express key components of the amyloidogenic cascade and can be used to explore potential treatments.[38] For example, carnosic acid (CA) reduced apoptosis in cells exposed to Aβ42, and blocking certain adenosine receptors helped prevent Aβ toxicity. These cell models help uncover the mechanisms of AD and aid drug development.[39]
Molecular simulation models
Researchers have recently developed molecular simulation models to mimic the molecular events in Alzheimer’s disease (AD) and accelerate drug development. One approach involved creating an NADPH oxidase-nitric oxide system to replicate the inflammatory cascade in AD, triggering neuronal death through an inflammatory process.[40] Additionally, molecular dynamics simulations of Aβ40 peptides showed that β-sheet breakers can prevent Aβ40 aggregation by stabilizing its structure. These models provide valuable insights into the molecular mechanisms of AD and allow for efficient screening of potential therapeutic compounds.[41]
CONCLUSION
Alzheimer’s disease is a chronic neurodegenerative condition that progresses over many years. Alzheimer’s disease (AD) is a major global health challenge due to its complex neuropathology, involving amyloid-beta (Aβ) aggregation, neuroinflammation, and cholinergic dysfunction. Despite extensive research, effective treatments remain limited. Current therapies provide modest benefits but come with side effects. In vitro models, including brain tissue cultures, stem cell-derived models, and molecular simulations, are crucial for studying AD and testing therapies. These models help explore key disease processes like Aβ accumulation and neuroinflammation, paving the way for targeted and personalized treatments. With AD’s growing prevalence, further research is essential to develop effective therapies and improve patient care.
REFRENCES




Shabeeha Mathapulan, Mridhul Mohan P.*, E. Tamil Jothi, Jeena Chandran N., Advances in Alzheimer’s Disease Research: Neuropathology, Therapeutic Strategies, and In Vitro Models, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 3, 1008-1015. https://doi.org/10.5281/zenodo.15013428
 10.5281/zenodo.15013428
10.5281/zenodo.15013428
