Modern Institute of Pharmaceutical Sciences, Indore
Molecular docking plays a vital role in advancing scientific research, particularly in drug development and understanding biomolecular interactions. This computational approach simulates the binding of small molecules, such as potential drugs, to specific biological targets like proteins or DNA. By predicting the optimal binding configuration and energy, molecular docking helps identify promising drug candidates and sheds light on the underlying mechanisms of biological processes. The accuracy of this method relies heavily on a robust scoring function, which distinguishes between strong and weak binding interactions, enabling the identification of potential medication candidates for validation in experiments. This in-depth review delves into the fundamentals and applications of molecular docking in the realm of drug discovery. It outlines the essential stages of drug design, including structure-based approaches, and explores the intricacies of molecular docking, encompassing various types, underlying principles, and algorithms. The review also examines the critical role of scoring functions in assessing binding affinities and discusses the challenges and limitations inherent in molecular docking. Furthermore, it highlights the significant impact of molecular docking on streamlining drug discovery, making it more efficient and cost-effective. Looking ahead, the review anticipates future advancements in docking algorithms, integration with complementary computational methods, and innovative approaches, which are expected to further augment the capabilities of molecular docking. The primary focus of this review is on the central concept of molecular docking, its diverse interactions, underlying principles, and applications, as well as the challenges, limitations, and available software for molecular docking.
Molecular docking is a powerful computational tool that plays a vital role in drug discovery by simulating the interactions between molecules and their targets. This structure-based approach enables researchers to identify promising new compounds with potential therapeutic benefits by predicting how ligands bind to specific targets at the molecular level. Moreover, it facilitates the understanding of structure-activity relationships without requiring prior knowledge of other modulators' chemical structures. The docking process involves a multi-step procedure to accurately predict the binding conformation and orientation of ligands within a target's binding site. The primary objectives of docking studies are to achieve precise structural modeling and accurately predict biological activity. This is accomplished through a combination of docking algorithms, which position small molecules within the active site, and scoring functions, which assess the interactions between compounds and targets, ultimately forecasting their biological efficacy [2]. This approach has found extensively used as a fast and cost-effective scholarly research environments and commercial industries over the past few decades. Despite its long-standing presence, there are still several challenging aspects that have yet to be resolved. One such challenge is the ability to accurately and easily identify true ligands from a set of molecules. Furthermore, pinpointing the exact three-dimensional arrangement of the ligand as it binds to a specific target molecule's active site continues to pose a significant challenge [3].
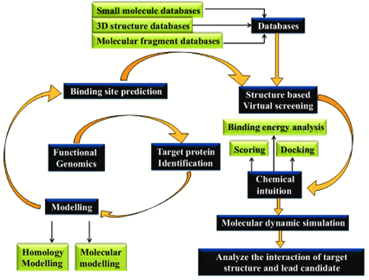
Figer no. 1
Drug Design:
The process of drug design is a dynamic and inventive field that harnesses insights into specific biological targets to create novel treatments. At its core, drug design involves crafting molecules with a precise shape and charge that perfectly complement their target, enabling a seamless interaction. In today's era of vast data, computer simulations and bioinformatics play a pivotal role in drug design. Notably, the field has expanded beyond small molecules to encompass biopharmaceuticals and therapeutic antibodies, which have become vital components. Advances in computational methods have significantly enhanced the effectiveness of these protein-based treatments, improving their binding affinity, selectivity, and stability. This ongoing progress is unlocking new pathways for the development of targeted and efficacious medications [4]. The journey of drug design and discovery involves a multifaceted approach, employing computational, experimental, and clinical models to identify promising new therapeutic agents. Despite significant strides in biotechnology, pharmacology, and our understanding of biological processes, the drug discovery process remains complex, time-consuming, and costly. The first step in drug design entails crafting molecules that mirror the shape and structure of their target, enabling precise binding. Modern drug design relies heavily on computer simulations and bioinformatics. It's essential to consider the synthetic feasibility, leveraging the inherent physio-chemical properties of the compounds involved. Furthermore, when developing synthesis methods, factors such as environmental sustainability, production safety, economic viability, and green chemistry principles must be carefully weighed [5].
Types of Drug Design:
Ligand- Based Drug Design
Structure- Based Drug Design
Ligand- Based Drug Design:
When structural information about the therapeutic target is scarce, an alternative strategy called Ligand-Based Drug Design (LBDD) comes into play. Unlike Structure-Based Drug Design (SBDD), LBDD doesn't necessitate prior knowledge of the target's mechanisms of action. Instead, it leverages structural data and bioactivity information from small molecules. This approach is rooted in the principle, first proposed by Hendrickson in 1991, that structurally similar molecules tend to exhibit similar properties. A pivotal step in LBDD involves curating and preparing libraries of small molecules. Common techniques employed in LBDD include similarity-based searches, quantitative structure-activity relationships (QSAR), and pharmacophore modeling. These methods facilitate the identification of potential drug candidates by exploiting their structural similarities and activity correlations.[6]
Structure- Based Drug Design:
The advent of Structure-Based Drug Design (SBDD) has revolutionized the process of designing and optimizing drug candidates by harnessing the power of three-dimensional structural information of target proteins. This approach relies on the precise geometric details of the target protein's 3D structure, typically elucidated through advanced techniques such as X-ray crystallography, NMR spectroscopy, or cryo-electron microscopy (cryoEM), to guide the design of novel therapeutics.[7] The widespread adoption of Structure-Based Drug Design (SBDD) by pharmaceutical companies and researchers has yielded significant breakthroughs, including the development of successful drugs like HIV1 protease inhibitors, raltitrexed, and norfloxacin. However, traditional SBDD methods, which involve manual modeling, custom scoring functions, and exhaustive enumeration, can be resource-intensive and time-consuming. To address these limitations, a novel approach called geometric deep learning has emerged. This innovative method seeks to revolutionize the SBDD process by harnessing the power of deep learning algorithms, promising to streamline and enhance drug discovery efforts.[7] "The advent of 3D protein structures has transformed the landscape of drug discovery, enabling a more precise and streamlined approach to structure-based drug design (SBDD). This method leverages the detailed molecular architecture of target proteins to guide the identification and optimization of lead compounds, yielding a more efficient and accelerated discovery process. By gaining insight into the molecular mechanisms of disease through 3D structural analysis, SBDD facilitates the rational design of potential therapeutics, allowing researchers to target specific biological pathways with greater accuracy and confidence. [6] The initial phase of a typical structure-based drug design (SBDD) workflow involves the identification and validation of a target protein. To elucidate the 3D structures of proteins with therapeutic potential, researchers employ experimental techniques such as NMR, X-ray crystallography, or cryo-electron microscopy, which are integral to integrative structure biology. In cases where experimental structures are unavailable, computational methods can be utilized to predict the protein's 3D architecture. Three prominent structure prediction approaches exist: comparative modeling, threading, and ab initio modeling, with homology modeling being a reliable and widely-used method. This approach leverages knowledge of homologous proteins with greater than 40% similarity to predict the target protein's 3D structure. Various validation methods are also available to ensure the accuracy of the predicted model. Once the target protein's structure is determined, the next step involves identifying the binding pocket, a small cavity where ligands interact with the target to produce the desired therapeutic effect.[8]
Process of Structure- Based Drug Design:
The structure-based drug design paradigm is revolutionizing the development of innovative therapeutics for a wide range of diseases. Breakthroughs in high-throughput protein structure determination are transforming the drug discovery landscape, enabling researchers to initiate the design process with a clear understanding of the protein's architecture. This approach facilitates the identification and optimization of novel ligands, tailored to interact with specific protein targets. The following overview aims to delineate the key stages involved in this rational drug design methodology.[9]
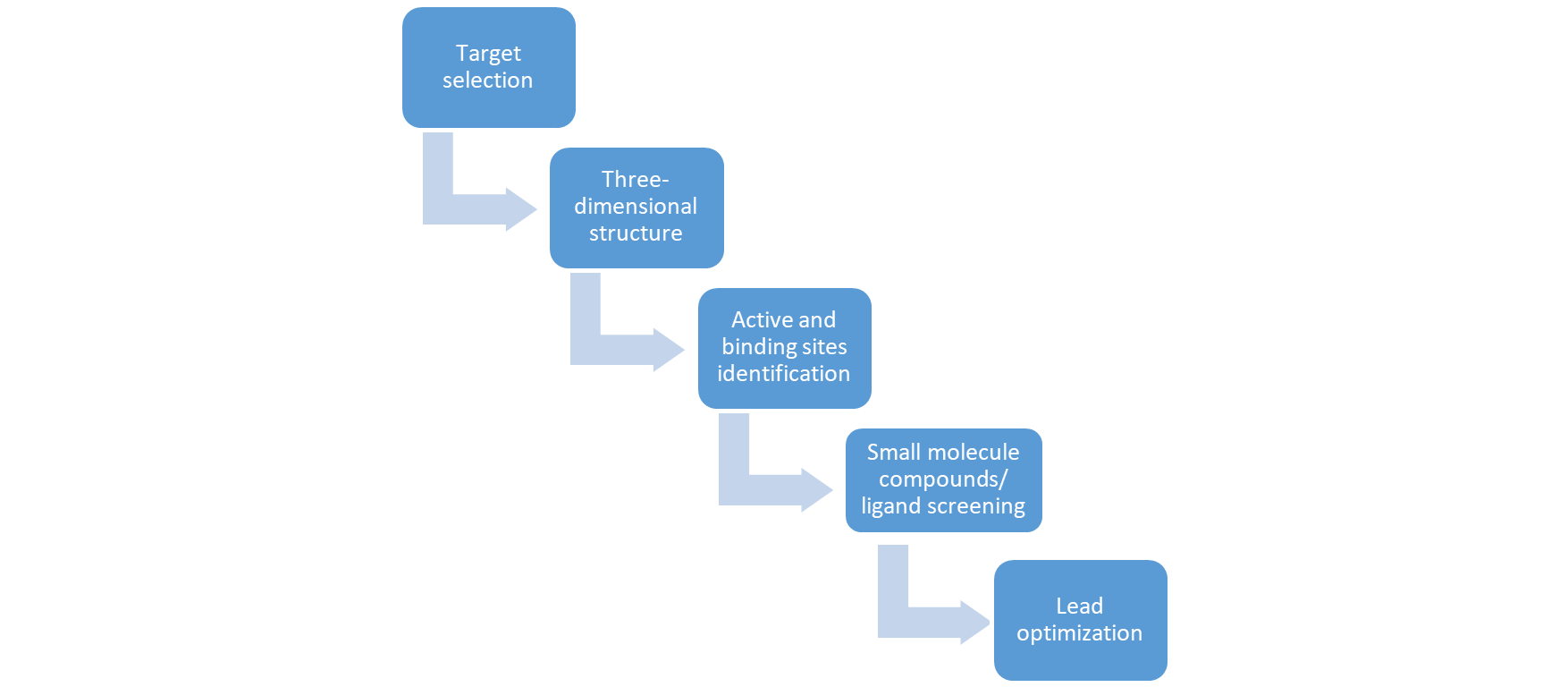
Steps Involved in Structure-based Drug Design
The identification of a drug target, a crucial macromolecule involved in disease pathology, is a multifaceted process that commences with a comprehensive understanding of its structural biology. To pinpoint a viable target, researchers consider a range of factors, including functional predictions, pathway analysis, disease correlations, and structural insights. By leveraging various bioinformatics tools, investigators can gather and integrate this information to reveal potential targets that play a pivotal role in disease mechanisms, ultimately informing the selection of a promising target for therapeutic intervention. [9]. When designing drugs to combat infectious diseases, the ultimate objective is to achieve complete eradication of the pathogen. To accomplish this, the target of the drug should be strategically located in a critical juncture of a metabolic pathway, such that its inhibition would be catastrophic for the pathogen's survival. Ideally, the target should be unique and non-redundant, ensuring that no alternative pathways can bypass the inhibitory effect. Additionally, specificity is paramount, where the drug's mechanism of action should be precisely defined, ensuring selective binding to the target protein and minimizing off-target effects [9].
Once a drug target has been identified, precise structural information is essential to elucidate its functional mechanisms. Understanding the three-dimensional architecture of a protein is crucial for deciphering its biological role. To acquire this critical information, researchers employ three primary methods to determine structural data, which are:
X-ray crystallography (XRC) is a cutting-edge technique that harnesses the power of X-rays to unravel the molecular structure of proteins. By diffracting X-rays through crystalline structures, XRC provides unparalleled high-resolution insights, making it a cornerstone of drug design. Once the intricate 3D architecture of a protein is revealed, computational tools utilize the precise atomic coordinates to reconstruct the protein's structure, enabling a deeper understanding of its functional mechanisms and paving the way for innovative drug development. [9] Nuclear magnetic resonance (NMR) spectroscopy is a powerful analytical tool that leverages the interaction between magnetic fields and electromagnetic waves to elucidate the molecular architecture of organic compounds. By probing the magnetic properties of atoms, NMR provides detailed information on the structure, dynamics, and interactions of both target molecules and ligands, offering crucial insights that inform drug discovery and development.[9] When the three-dimensional structure of a target molecule remains unknown, homology modelling offers a powerful solution. This computational technique leverages available sequence data and relies on a structurally characterized template sharing significant sequence similarity (at least 30%) with the target. By harnessing this approach, researchers can generate accurate and reliable three-dimensional models of protein structures, facilitating a deeper understanding of their architecture and function. As a cornerstone of structural biology, homology modelling plays a vital role in elucidating protein structures and informing drug discovery efforts. [9].
Once the target's structure is elucidated, the next step involves pinpointing the binding site, a recessed region with specific chemical properties, including hydrogen bond donors and hydrophobic features. This site serves as the docking point for a small molecule, known as a ligand, which can be administered orally. When a ligand successfully binds to the target at this site and demonstrates inhibitory activity, it is deemed a lead compound or lead candidate, marking a crucial milestone in the drug discovery journey [9]
The core focus of computational drug design is to investigate the binding interactions between inhibitors and their targets. After pinpointing potential binding sites on a target molecule, researchers employ a combination of computational and experimental approaches to identify and evaluate promising ligands that can effectively bind to and inhibit the target. Although computational methods for structure-based drug design are still evolving and not yet fully developed, ongoing advancements in algorithms and software are continually emerging. The field can be broadly categorized into three key areas: De Novo design, molecular Docking, and Scoring techniques, which collectively drive the discovery of novel leads and the optimization of existing ones [9]. De novo design is a cutting-edge approach that involves crafting novel ligands with high affinity for a specific receptor. To address the complex challenge of ligand binding, researchers typically break it down into two interconnected components: docking and scoring. Docking entails predicting the optimal binding pose of a ligand to its receptor, given their known structures. Scoring, on the other hand, involves assessing the binding affinity of the ligand based on its predicted bound configuration. These complementary methods work in tandem to facilitate the discovery of potent drug molecules, enabling researchers to tackle the intricacies of structure-based drug design with greater precision and accuracy [9].
After successfully identifying the ligand, it is referred to as the lead compound. Before moving on to preclinical trials, the lead compound needs to undergo evaluation for its binding affinity. This evaluation process involves assessing various factors such as bioavailability, oral viability, chemical and physical stability, as well as ease of production.[9]
Success stories in structure-based drug design include:

Docking:
Docking is a virtual simulation technique used to study molecular interactions. With its exceptional precision in forecasting ligand conformation and binding modes within a target's active site, this technique has emerged as the gold standard in Structure-Based Drug Design (SBDD), earning its widespread adoption and acclaim [8]. Docking is commonly used to modulate the affinity and activity of small molecules by predicting how they align with specific target molecules. Docking research serves as a vital tool in elucidating the structural makeup of potential therapeutics, with the primary goal of achieving an optimal energetic state by precisely aligning and configuring the binding agent and enzyme, thereby minimizing the system's overall free energy. By doing so, it helps in understanding and improving the interactions between the two entities, ultimately aiding in the development of effective therapeutic treatments.[10] In the pursuit of novel therapeutics, docking has become an indispensable tool in Structure-Based Drug Design (SBDD), allowing researchers to visualize and analyze the binding patterns and energies of receptor-ligand complexes. As the field has matured, a staggering array of docking programs has emerged, providing researchers with a wealth of choices – over 50 and counting – to tailor their approach to the specific needs of their research. While there are no prescribed tools for SBDD, each software utilizes different algorithms and scoring functions, resulting in varied docking results.[11] Molecular docking offers a powerful framework for elucidating drug-biomolecular interactions, enabling the rational design and discovery of novel therapeutics. By virtually inserting a ligand into a specific binding site on a DNA or protein target, researchers can predict the formation of a stable, non-covalent complex with enhanced specificity and potential efficacy. This technique is particularly valuable when the three-dimensional structure of the target protein is known, making it a cornerstone of virtual screening approaches.[12] Discovering the most probable binding conformations entails a dual-phase approach. First, a vast conformational landscape is explored, encompassing numerous potential binding orientations. Next, the interaction energy of each predicted conformation is accurately predicted. Molecular docking software facilitates this process through a cyclical evaluation, employing scoring functions to assess ligand conformations. This iterative process is repeated until a minimum energy state is achieved, signifying an optimal binding conformation [13].
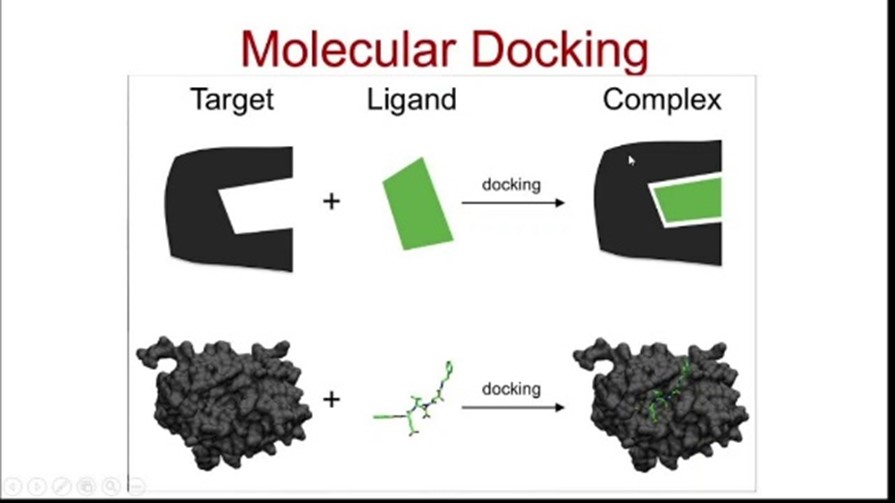
Molecular docking involves various types of interactions, including:
Types of Docking:
Docking Software:
Molecular docking has become an essential crucial assest that greatly boosts the effectiveness of high-throughput computer-generated screening methods used in both academics institutes and pharmaceutical drug discovery processes. Over the years, major breakthroughs in computer technology, advanced software and platforms, and enriched publicly accessible chemical databases have greatly enhanced the accuracy, efficiency, and value of computational screening approaches. As a result, numerous docking software programs have been developed for research and commercial applications. Additionally, a significant upsurge has been observed increase in the number of research papers concentrating on "docking" over the past two to three decades. These developments have revolutionized the field and have greatly facilitated the process of drug screening and discovery.[10]
There are some Docking Software:
PRINCIPLES OF DOCKING
Fundamental Principles in Molecular Docking:
Initiate the process by acquiring the three-dimensional structure of the target protein (receptor), either through experimental determination or computational prediction using techniques such as homology modeling. Next, refine the protein structure by removing superfluous water molecules and co-crystallized ligands. Finally, prepare the ligand molecule by obtaining its three-dimensional configuration and optimizing its geometric parameters to ensure a precise representation.
Proceed by locating the specific region on the protein where the ligand is likely to interact. This can be achieved by leveraging existing experimental data or employing computational methods that detect potential binding cavities. These tools help pinpoint the most probable binding site, paving the way for further analysis. While computational tools use calculations to predict binding sites based on factors like shape and charge. Defining the binding site helps us focus on the right area for the docking study, ensuring accurate results.
To explore the different ways the ligand can bind to the protein, we Create multiple conformations or flexible forms of the ligand to mimic its adaptability. This can be done using techniques like molecular dynamics simulations or conformational searching algorithms. These methods allow us to examine various binding modes and understand the ligand's flexibility within the binding site. By exploring different conformations, we can uncover the most favorable interactions between the ligand and the protein.
To choose the right docking algorithm, Select a suitable docking approach based on the problem's complexity and available computational resources, choosing from options like rigid docking (for static ligand-receptor interactions), flexible docking (for dynamic ligand or receptor conformations), or hybrid methods that integrate multiple strategies. Each algorithm has its strengths and limitations, so pick the one that suits your needs. This step ensures accurate predictions of ligand-protein binding interactions.
In this phase, the ligand is methodically placed and aligned within the receptor's binding site, allowing the docking algorithm to assess and rank various ligand conformations. The evaluation is based on key factors such as spatial compatibility, hydrogen bonding potential, electrostatic complementarity, and hydrophobic interactions. The ultimate goal is to pinpoint the ligand pose that exhibits the strongest predicted binding affinity, indicating a stable and favorable interaction with the receptor.
After the docking simulation, the next step is to Evaluate the docking results and prioritize the ligand poses according to their predicted binding strengths or scoring functions. This is done using scoring functions, which help Evaluate the intensity of ligand-receptor binding and identify the strongest probable binding configuration. By evaluating and comparing the scores of different ligand poses, we can determine which poses are more likely to have a favorable binding affinity with the receptor. This analysis allows us to prioritize and select the ligand poses that show the highest potential for effective binding.
After the docking simulation, the results are analyzed and ligand poses are ranked based on their predicted binding strengths. The docking results can be validated through comparison with empirical data, if applicable. This helps evaluate the reliability of the predicted results. If needed, the ligand-receptor binding entities can be further refined using post-docking refinement techniques, including molecular dynamics and energy minimization, to fine-tune the accuracy of the predicted binding conformation.
Once the docked conformations are obtained, they are analyzed to elucidate the mechanisms of ligand-receptor binding. This analysis involves identifying key residues involved including key aspects such as hydrogen bonding networks, hydrophobic contacts, and other crucial factors that influence the ligand's binding strength. By understanding these interactions, we can gain a more profound insight into the molecular recognition mechanism. This information can then guide further experimental or computational studies, helping to refine and optimize the ligand-receptor interactions.
To validate and support the findings of the docking results, it is important to integrate them with experimental data and other computational methods. This integration can necessitate comparing the contrasting docking results with empirical binding affinity data conducting structure-activity relationship (SAR) evaluation, or even uncovering innovative chemical scaffolds for lead development. By combining different sources of data and information, we can strengthen the reliability and robustness of the findings, ultimately guiding further research and development in the field of drug discovery.
After conducting the molecular docking study, it is important to summarize the findings and examine their consequence within the framework of the research inquiry or aims. This involves highlighting the key findings and their significance in addressing the research question or achieving the objectives of the study. It is also important to acknowledge the power and limitations of the molecular docking approach. By recognizing its strengths and weaknesses, we can provide a balanced assessment of the method. Additionally, it is valuable to suggest potential avenues for future research or applications of molecular docking, suggesting potential areas for further research and development [14]
Different Types of Molecular Docking Algorithms:
Algorithms for Rigid Body Docking are used in docking simulations where both the binding molecule and target protein are assumed to remain fixed. These algorithms examine the ligand's conformational flexibility within the binding site, while maintaining a static receptor.
Methods that Allow for Conformational Flexibility are designed to incorporate conformational adaptability in the ligand, receptor, or both during the docking simulation. When considering ligand flexibility, these algorithms generate multiple conformations of the ligand to examine its various possible conformations shapes and orientations within the receptor's active site. On the other hand, receptor flexibility can be accounted for by permitting movements of side chains or the main chain within the receptor's binding site.
Induced-fit docking algorithms are specifically designed to simulate the structural changes that take place in both the ligand and receptor upon complex formation. These algorithms utilize a dual-step strategy: first, a two-stage approach is employed, where the first stage involves rigid-body docking, followed by a refinement stage that introduces flexibility in both molecules. This allows for a more accurate representation of the dynamic nature of ligand-receptor interactions.
Hybrid docking approaches are designed to enhance the accuracy of docking predictions by combining multiple docking strategies. These approaches often merge rigid, flexible, and induced-fit docking techniques to create a more robust and effective docking protocol strike a balance between speed and accuracy in docking simulations.
Ligand-based docking methods take a different approach by not relying on the explicit structure of the receptor. Instead, they tap into the wealth of information from known ligands or ligand databases to forecast binding modes. By leveraging techniques like molecular fingerprints, pharmacophore modeling, or shape-based matching, these methods can predict how ligands will interact with the receptor. This approach proves especially valuable when the target protein's structure remains elusive or challenging to determine, providing a powerful alternative for understanding receptor-ligand interactions. By leveraging the characteristics of known ligands, these methods provide valuable insights into potential binding interactions and aid in the identification of novel therapeutic compounds.
Ab initio docking algorithms are specifically designed to predict the binding mode de novo, without relying on existing structural information or ligand data. These algorithms employ utilizing fundamental physical principles, energy computations, and optimization technique to explore sampling the conformational flexibility of both molecules to identify the most stable docking configuration. Ab initio methods require significant computational resources and are generally limited to smaller molecular system.
Machine learning-based docking methods are gaining popularity in the field of molecular docking as they offer improved accuracy and speed. These algorithms utilize machine learning techniques by training on a a dataset of experimentally determined ligand-receptor complexes. which trains the models are then used to forecast the binding strength or docking orientations of novel ligands. By leveraging the power of machine learning, these these methods can augment conventional techniques and improve the effectiveness of virtual screening. [14]
Scoring Functions and their Role in Evaluating Binding Affinities:
The Auto Dock scoring function adopts a holistic approach to predict binding affinity, integrating empirical terms that model various molecular interactions. These terms include van der Waals interactions, electrostatic interactions, hydrogen bonding, and desolation energies. These factors help determine how well a ligand binds to a receptor, aiding in virtual screening and drug discovery.
X-Score assesses molecular binding through a 5-term empirical scoring function, covering van der Waals, hydrogen bonding, electrostatics, hydrophobicity, and solvation effects. It helps evaluate the strength of molecular binding between a ligand and a receptor in drug discovery and virtual screening.
ChemScore estimates binding affinity by considering hydrophobic contacts, hydrogen bonds, enthalpic contributions, and metal-ligand interactions. It helps assess the affinity between a ligand and its receptor in drug discovery and virtual screening.
Physics-based scoring functions use advanced methods like molecular mechanics, quantum mechanics, and molecular dynamics simulations to calculate binding affinity in a more detailed and accurate way. They provide glimpse into the structural arrangement and dynamic aspects of ligand-receptor interactions, aiding in drug discovery efforts.
MM-PBSA is a computational approach that estimates the binding affinity energy by combining molecular mechanics and continuum electrostatics. It accounts for various energy components, including van der Waals, electrostatic, solvation, and entropic effects. This approach provides a comprehensive analysis of the binding interactions, helping researchers understand the stability and strength of ligand-receptor complexes.
MM-GBSA is a variant of MM-PBSA that employs a generalized Born approach for solvation energy calculations, rather than the Poisson-Boltzmann method. It's another method to calculate binding free energy in a slightly different way.
Prime, which is part of the Schrodinger suite, is a physics-based scoring function his approach merges molecular mechanics force fields with a continuum solvent model to provide an estimate of the ligand-receptor binding energy.[14]
Application of docking in drug discovery
In silico screening, powered by molecular docking, is a pivotal strategy in drug discovery, enabling the exploration of vast chemical libraries to uncover potential therapeutic leads that bind to a specific protein target, with docking simulations providing valuable insights into binding strength and orientation.
Molecular docking plays a crucial role in lead optimization. It helps evaluate and refine lead compounds, improving their ability to bind selectively and with high affinity to the target protein. Docking simulations inform medicinal chemists on how to tailor the chemical structure of lead compounds to strengthen interactions and refine their pharmacological profile.
Docking is instrumental in predicting The ways in which small molecules interact with their target protein's active site. It reveals the precise molecular contacts and crucial amino acid residues that participate in ligand-protein binding, essential for understanding the underlying mechanism and developing novel therapeutic agents.
Docking simulations facilitate structure-activity relationship (SAR) studies by investigating the binding properties and molecular interactions of related compounds, enabling researchers to pinpoint key structural elements that influence binding affinity and inform the design of enhanced compounds.
Docking is a cornerstone of fragment-based drug discovery (FBDD), where it's used to screen small molecular fragments for binding to target proteins, revealing key interaction hotspots and seeding the development of initial lead compounds. Docking then informs the iterative assembly and refinement of these fragment hits into more comprehensive, drug-like molecules through strategies such as fragment elaboration, combination, or extension.
Docking is a valuable tool for investigating and manipulating protein-protein interactions (PPIs), focusing on specific binding sites or interfaces within protein complexes. By discovering small molecules that either disrupt or stabilize PPIs, docking provides potential therapeutic approaches for diseases characterized by abnormal PPIs.
De novo drug design leverages docking to create novel compounds in silico, tailoring them to meet specific design criteria and constraints. This approach enables the evaluation of compound fitness and binding affinity within the target protein's active site, facilitating the identification and refinement of promising drug candidates.
Docking facilitates drug repurposing by virtually screening approved drugs or compounds with established properties against novel targets, uncovering potential off-target interactions or new therapeutic applications for existing medications. [14]
CHALLENGES AND LIMITATIONS
The application of docking tools and interpretation of their results pose significant challenges. Each program has its unique set of limitations and shortcomings, leading to variability in output quality and reliability. Moreover, the software's performance may degrade when handling complex chemical structures that surpass its design capabilities.[15] Ongoing research efforts are focused on overcoming the existing limitations and developing innovative methodologies to augment the precision and utility of molecular docking in drug discovery.
Docking studies face a significant challenge in the accuracy of scoring functions. The accuracy of these functions is limited by their reliance on simplified energy models and assumptions, which may not adequately account for the intricate dynamics of ligand-protein binding. Ongoing research is focused on improving the accuracy of scoring functions to better predict binding affinities. Further advancements are needed in this area to enhance the reliability of docking results.
Molecular docking methods typically only scratch the surface of the conformational landscape, potentially overlooking crucial binding modes and neglecting the dynamic nature of ligands. However, exhaustively exploring this vast space is a daunting task, hindered by computational constraints and the astronomical number of possible conformations.
Incorporating solvent effects is vital for precise docking predictions, but simulating solvents accurately, particularly in complex protein-ligand systems, poses significant computational challenges. Oversimplified solvation models employed in docking may fail to capture the intricacies of solvent interactions, leading to errors in binding affinity predictions.
Proteins and ligands exhibit flexibility and dynamics that can affect their binding interactions, but many docking methods fail to consider this by treating proteins as static entities. Incorporating protein flexibility and dynamics into docking simulations is an active research area, but it requires significant computational resources.
Effectively exploring the conformational space of ligands, particularly those with numerous rotatable bonds, can be a daunting task. Inadequate sampling of ligand flexibility may lead to missed binding opportunities and flawed predictions.
Molecular docking predictions may be less reliable for novel targets or binding sites with scarce structural data, as the accuracy of docking relies heavily on knowledge of the target protein's structure and binding site. To overcome this limitation, techniques such as homology modeling or comparative modeling can be employed, but they introduce uncertainties in the predicted protein structure, potentially affecting docking outcomes.
Molecular docking calculations can be computationally intensive, particularly for complex protein-ligand systems or exhaustive conformational searches. Sufficient computational power and time are essential for thorough docking analyses, but there are practical limits to the scope and scale of calculations that can be accomplished within reasonable timeframes.
The tendency of molecular docking methods to overestimate ligand binding affinities can lead to a higher incidence of false positives. This limitation stems from the simplification of energy models, neglect of entropy factors, and difficulties in precisely accounting for solvation influences. [14]
FUTURE PERSPECTIVES AND ADVANCES
Molecular docking plays a vital part in the discovery of new drug, offering valuable opportunities. However, its prediction performance is limited due to inherent factors. To address this, docking is now often combined with alternative computational approach such as Ligand-centric methods, receptor-centric methods, quantum mechanics, machine learning, and artificial intelligence (AI). This integration helps overcome some of the significant limitations of docking, making it a more robust platform for rational drug design. [15] Frequently used computational approaches that enhance molecular docking:
MD simulations can further refine the protein-ligand complex generated by molecular docking. By mimicking the dynamic behavior of the complex over time, MD simulations offer valuable insights into the binding interaction's stability, protein and ligand flexibility, and binding free energy, providing a more comprehensive understanding of the complex's dynamic properties and refining the predicted binding modes.
QM methods can be applied to investigate specific regions of the protein-ligand complex that demand precise quantum mechanical analysis. For instance, QM calculations can deliver detailed insights into the electronic properties and chemical reactions within the active site. By combining QM calculations with molecular docking, researchers can achieve a more profound understanding of intricate systems, uncover reaction mechanisms, and enhance binding affinity predictions.
When experimental structures of the target protein are unavailable, homology modeling offers a viable alternative. By combining homology modeling with molecular docking, researchers can create reliable 3D models of the target protein and conduct docking simulations, facilitating the identification of potential binding sites, prediction of binding modes, and optimization of lead compounds
By combining machine learning and AI with molecular docking, researchers can significantly enhance the precision and efficiency of predictions. For instance, machine learning algorithms can be trained on extensive datasets of known protein-ligand interactions to forecast binding affinity, evaluate ligand efficacy, and identify top candidates for experimental testing. These models can augment docking results, offering additional perspectives on ligand-target interactions. [14]
CONCLUSION:
In summary, molecular docking has emerged as a groundbreaking technology in drug discovery, allowing researchers to forecast and dissect the intricate relationships between small molecules and target proteins. Its contribution to expediting the drug development process is immense, offering unparalleled understanding of binding affinity and mechanism of action for prospective drug candidates. "Molecular docking enables the prediction of an ideal ligand orientation on its target protein, as well as the identification of various binding modes within the target's binding site. This capability facilitates the design of drug candidates with enhanced potency, selectivity, and efficacy. The integration of docking with scoring functions enables the virtual screening of vast databases to identify promising drug candidates that can selectively target molecules of interest. Molecular docking is instrumental in predicting the initial binding properties of drugs to nucleic acids, revealing a crucial correlation between a drug's molecular architecture and its potential cytotoxic effects. After conducting thorough evaluations of the target, ligands, and docking method efficacy, molecular docking is utilized to forecast binding affinities with high accuracy. The flexibility of ligands is effectively addressed, posing minimal challenges. Molecular docking plays a pivotal role in hit identification, enabling researchers to pinpoint potential drug candidates by predicting their binding affinity to targeted proteins or receptors.
REFERENCE





Yadav Khushi , Joshi Ankur , Khemani Purva, Malviya Sapna, Kharia Anil , A Short Review Docking: Structure Based Drug Design, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 9, 1013-1027. https://doi.org/10.5281/zenodo.13820440
 10.5281/zenodo.13820440
10.5281/zenodo.13820440
