DR. Naikwadi College of Pharmacy Jamgaon, Sinnar 422102.
High-Performance liquid chromatography (HPLC) is widely recognized as the most commonly used separation technique for the detection, separation, and quantification of drugs. To optimize HPLC methods, various chromatographic parameters are evaluated, including sample pretreatment, choice of mobile phase, column selection, and detector type. This article aims to discuss the processes involved in method development, optimization, and validation. The development and validation of analytical methods are critical in the discovery, development, and manufacturing of pharmaceutical drugs, as well as in research involving humans and animals. HPLC is a dominant technique in separation science, where analytes are separated by passing through a column packed with micrometre-sized particles. To comply with good manufacturing practices (GMP), pharmaceutical industries are required to establish comprehensive validation policies outlining the approach to validation. This article focuses on the optimization of HPLC conditions and provides an overview of the introduction, principles, types, instrumentation, method development and validation processes in HPLC. The validation of HPLC methods provides critical information about various parameters such as range, linearity, specificity, accuracy, precision, limit of detection (LOD), limit of quantification (LOQ), robustness, ruggedness and system suitability. These validation processes must adhere to regulatory guidelines, such as those outlined by the International Council for Harmonisation (ICH).
High performance liquid chromatography (HPLC) is a highly effective tool in analytical chemistry, widely used for separating, identifying, and quantifying compounds in any liquid-dissolvable sample. It is considered one of the most precise methods for both qualitative and quantitative analysis of drug products [1]. The technique operates by injecting a sample solution into a column filled with a porous stationary phase, while a liquid mobile phase pumped through the column under high pressure. Separation occurs due to differences in migration rates, which result from the varying partitioning of sample components between the stationary and mobile phases. Each component elutes at a distinct time based on its partitioning behaviour [2]. In HPLC, a compound with greater affinity for the stationary phase moves more slowly and covers a shorter distance, whereas a compound with lower affinity travels faster and cover a greater distance [3]. The primary goal of the HPLC method is to separate and quantify the main drug, reaction impurities, synthetic intermediates, and degradation products. HPLC has become one of the most advanced tools in analytical chemistry, capable of separating, identifying, and quantifying compounds in any liquid-dissolvable sample. It is widely recognized as one of the most reliable methods for both qualitative and quantitative analysis of drug products and for assessing their stability [4].

Figure no. 1 HPLC System
Principles:
The separation principle in both normal-phase and reverse-phase HPLC modes is based on adsorption. When a mixture is injected into the HPLC column, the components move at different rates depending on their relative affinities for the stationary phase. Components with higher affinity for the adsorbent move more slowly, while those with lower affinity travel faster. Since each component exhibits a unique level of affinity for the stationary phase, they are effectively separate.
Classification:
Normal phase mode:
The stationary phase exhibits polar characteristics, whereas the mobile phase is non-polar.
Reverse phase:
The stationary phase is non-polar, whereas the mobile phase is polar.
Adsorption chromatography:
The separation of the components is due to the change in the affinity of the compounds to the stationary phase.
Ion exchange chromatography:
An ion is used to separate a mixture of like charged ions.
Size exclusion or gel permeation chromatography:
A mixture of components of different molecular sizes is separated with gels that act like a sieve.
Isocratic separation:
In this method, the identical combination of mobile phase is utilized consistently during the separation procedure.
Gradient separation:
In this method, a mobile phase composed of lower polarity or elution strength is initially utilized, with a gradual increase in polarity or elution strength thereafter.
Analytical HPLC:
In this method, only the analysis of the sample is conducted, without performing sample retrieval.
Preparative HPLC:
In this method, the separate components of pure substance can be gathered with the assistance of a fraction collector.
Qualitative analysis:
It is employed for the purpose of determining the compound, identifying any impurities present, and determining the total number of components within the sample.
Quantitative analysis:
This process is used to measure the amount of each component in a mixture by comparing the peak areas of the standard and the sample [40].
Instrumentation of HPLC:
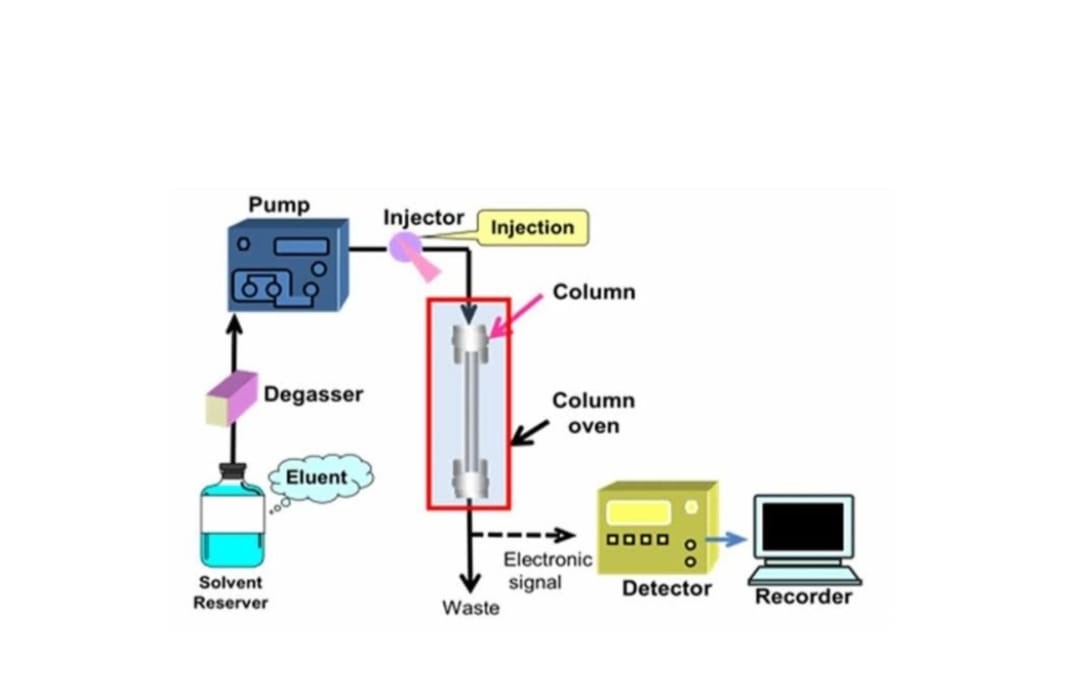
Figure no. 2 Instrumentation of HPLC
Components of the HPLC System,
Solvent Reservoir, Mixing Systems, Degassing System
The solvent is kept in the solvent reservoir, which is the mobile phase container. These reservoirs are typically made of glass or stainless steel that does not change colour. Glass bottles are the most commonly used type of solvent reservoir [5]. In addition to dispensing the mobile phase, the pump must also accurately and precisely mix solvents. There are two types of mixing units: low pressure mixing and high pressure mixing [6]. The process of removing air bubbles from the solution is carried out through the degassing mechanism, which utilizes ultrasonication and filtration methods for effective results [5].
High – Pressure pump
A pump’s function is to move liquid at a specific speed. Millilitres per minute (ml/min) is the unit of measurement for this speed. A common flow rate is between 1-2 ml/min. The pump’s pressure range is 6000-9000 psi (400-600 bar) [7]. Various types of pumps, such as continuous pressure pumps, pumps with syringe mechanisms, and pumps with reciprocating piston systems, are frequently employed in various applications [8].
Sample Injector
The sample injector is responsible for introducing the liquid sample into the mobile phase, while a sample valve is positioned between the pump and the column [5]. An auto sampler, known as an injector, has the capability to introduce the sample into the continuously moving mobile phase stream, which then carries it to the HPLC column. It is common for a standard sample volume to range from 5 to 20 microliters [7]. Manual injectors and automatic injectors represent the two categories of injectors [8].
Column
A column serves as the specific location for the separation of components to occur, with stainless steel being the material of choice for its construction. Its dimensions typically range from 5 to 25 centimeters in length and 2 to 4.6 centimeters in diameter on the inner side [7].
Detector
The detector has the capability to recognize and transform the different elements that flow out of the column into an electrical signal [7]. Two types of detectors are utilized in this context: specific detectors and bulk property detectors. Examples of specific detectors include UV-VIS detectors, photo diode array detectors, fluorescence detectors, and mass spectrometric detectors. On the other hand, examples of bulk property detectors encompass refractive index detectors, electrochemical detectors and light scattering detectors [8].
Data Recording system
The results are captured in a series of high points, and the computer linked to the screen automatically computes the space under each high point [9].
HPLC Method Development:
Developing and validating analytical methods are essential steps in the discovery, development, and production of pharmaceuticals. These methods are employed to confirm the identity, purity, strength, and effectiveness of pharmaceutical products. There are many factors to take into account when developing methods. For UV detection, they begin by collecting information on the analyte’s physicochemical properties (such as pka, log P, and solubility) to determine the most appropriate detection mode for analysis. Most of the analytical development work focuses on validating an HPLC method to indicate stability. The HPLC method aims to separate and quantify the primary active drug, any reaction impurities, all accessible synthetic intermediates, and any degradants [10,11]. When no established methods are available, new methods are developed to analyze novel products. Novel methods are developed to analyze existing pharmacopoeial and non-pharmacopoeial products, aiming to enhance precision and ruggedness while reducing costs and saving time. These methods are refined and validated through experimental trials. Alternative methods are suggested and implemented to replace the current procedure, with a comparison of laboratory data highlighting their respective advantages and disadvantages [12]. The factors driving the development of new drug analysis methods are:
Steps in HPLC method development include:
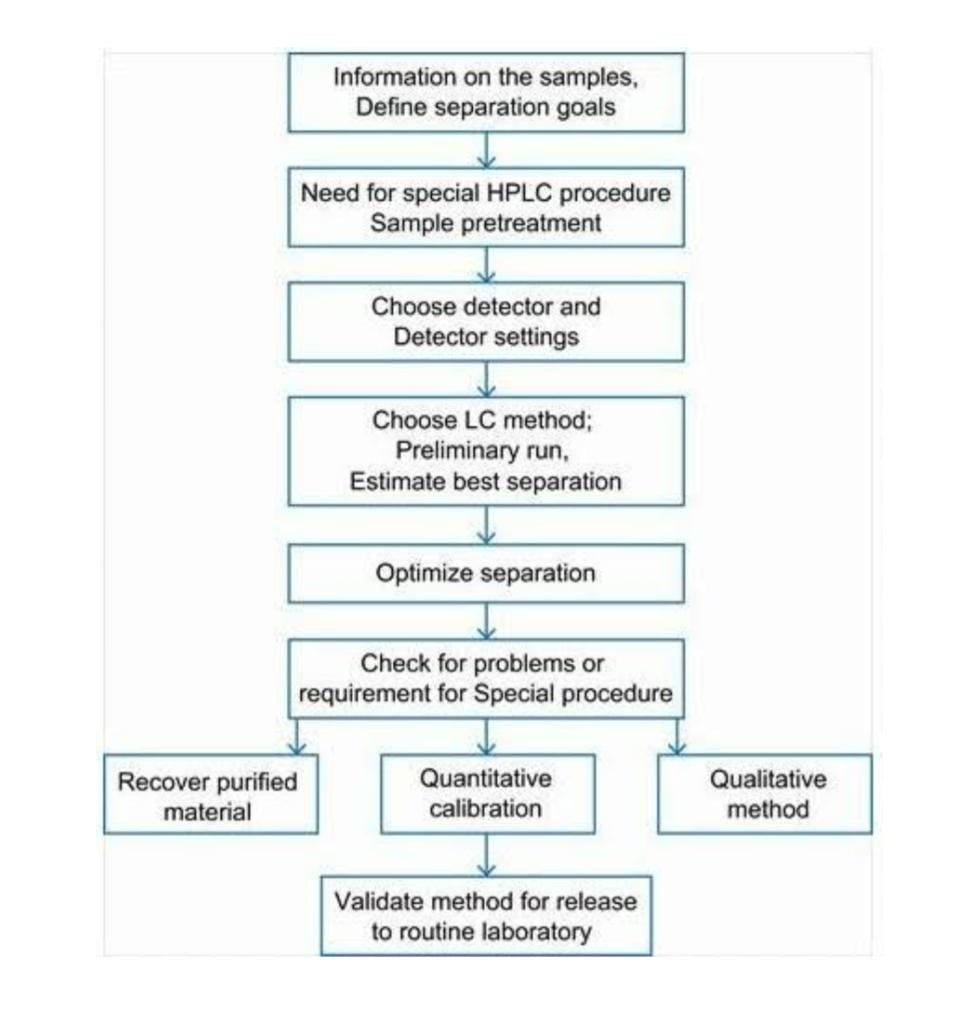
Figure no. 3 Steps in HPLC method development include
The physicochemical properties of drug molecule play a crucial role in method development. To design an effective method, it is essential to first examine the drug’s physical characteristics, including solubility, polarity, pka, and pH. Polarity is a physical property of a compound that helps an analyst determine the appropriate solvent and mobile phase composition. Molecular polarity can also be used to explain solubility behaviour. Polar solvents, like water, and nonpolar solvents, such as benzene, are immiscible. Generally, “like dissolves like,” meaning substances with similar polarities tend to dissolve in each other. The analyte’s solubility affects the selection of mobile phase or diluents. The analyte should be soluble in the diluents and must not react with any of its components. In HPLC method development, pH, and pKa values are critical factors. pH is defined as the negative logarithm (base 10) of the hydrogen ion concentration.
pH is defined as -log10[H3O+].
Selecting an appropriate pH for ionizable analytes often produces sharp and symmetrical peaks in HPLC. In quantitative analysis, these sharp, symmetrical peaks are essential for achieving low detection limits, minimal relative standard deviation between injections, and consistent retention times [14,15].
In the initial stages of method development, a set of starting conditions (detector, column, and mobile phase) is selected to produce the first “Scouting” chromatograms of the sample. Typically, reverse-phase separations on a C18 column with UV detection are employed. At this stage, it is necessary to decide whether to proceed with developing an isocratic or a gradient method.
Selection of column:
The column is undoubtedly the most crucial component of a chromatographic system. Selecting the appropriate column can lead to effective chromatographic separation, ensuring accurate and reliable analysis. Conversely improper use of a column can result in unclear, inadequate, and poor separations, yielding invalid or hard-to-interpret results [16]. The column is the central component of an HPLC system. When developing a method, altering the column has the most significant effect on the resolution of analytes. The ideal column for a particular application should consider factors such as the chemistry of the stationary phase, retention capacity, particle size, and column dimensions. An HPLC column consist of three main components: the hardware matrix, and stationary phase. Matrices such as silica, polymers, alumina and zirconium are used to support the stationary phase, with silica being the most commonly used. Silica matrices are durable, easily derivatized, manufactured with consistent spherical sizes, and resistant to compression under pressure. To select the ideal column for an application, it’s essential to consider the stationary phase chemistry, retention capacity, particle size, and column dimensions. An HPLC column consists of three primary components: the hardware, the matrix, and the stationary phase. Silica, polymers, alumina, and zirconium are among the matrices used to support the stationary phase in HPLC columns. Silica is the most commonly used matrix due to its strength, ease of derivatization, uniform sphere size production, and resistance to compression under pressure. Silica is chemically stable in most organic solvents and low-pH systems. However, drawback of using silica as a solid support is that it begins to dissolve at pH levels above 7. In recent years, silica-supported columns designed for high-pH applications have been developed. The properties of silica, including its structure, shape, and particle size, contribute to effective separation. Smaller particles lead to a higher number of theoretical plates. The type of stationary phase dictates whether a column is suitable for normal-phase or reverse-phase chromatography. Normal-phase chromatography utilizes a polar stationary phase and a non-polar mobile phase. Generally, polar compounds elute more slowly than non-polar compounds. The most common reverse-phase columns and their applications are outlined below. Propyl (C3), butyl (C4), and pentyl (C5), phases are suitable for ion-pairing chromatography (C4), peptides with hydrophobic Residues, and other large molecules. Compared to C8 or C18 phases, C3-C5 columns generally exhibit lower retention for non-polar solutes. Examples include Zorbax SB-C3, YMC-Pack C4, and Luna C5. In general , these columns are less resistant to hydrolysis than those with longer alkyl chains. Octyl (C8, MOS) phases have a broad range of applications. While slightly less retentive than C18 phases, they are still effective for analyzing pharmaceuticals, nucleosides, and steroids. The selection of the stationary phase or column is the first and most critical step in method development. A stable, high-performance column is essential for creating a robust and reproducible method. To prevent issues with inconsistent sample retention during method development, the column must demonstrate stability and reproducibility. Separation selectivity for specific components can differ not only between columns from different manufacturers but also between batches produced by the same manufacturer. Key factors influencing this variation include column dimensions, silica substrate properties, and the characteristics of the bonded stationary phase. Due to its diverse physical properties, silica-based packaging is the preferred choice for most HPLC columns today [17].
Selection of chromatographic mode:
Chromatographic modes are influenced by the molecular weight and polarity of the analyte. All case studies will focus on reversed-phase chromatography (RPC), the most widely used mode for small organic molecules. Ionizable compounds (acids and bases) are often separated by RPC using buffered mobile phases (to prevent the analyte from ionizing) or ion-pairing reagents [18].
Optimization of Mobile phase:
Buffer selection:
Various buffers, such as potassium phosphate, sodium phosphate, and acetate, were evaluated for system suitability parameters and overall chromatographic performance. After conducting sequential trials with these buffers, potassium dihydrogen phosphate was identified as the most effective for achieving optimal peak separation. Buffer concentrations of 0.02M, 0.05M, and 0.1M were tested, with no significant impact observed on the elution pattern or resolution. However, the 0.05M concentration notably enhanced the sensitivity of the method [19].
Effect of pH:
For ionizable analytes, selecting the appropriate mobile-phase pH relative to the analyte’s pKa is crucial to ensure the target analyte remains in its ionized or neutral form. Adjusting the mobile phase pH is a highly effective tool in the chromatographer’s repertoire, enabling simultaneous optimization of retention and selectivity for critical component pairs [19].
Effect of organic modifier:
In reverse-phase HPLC, choosing the type of organic modifier is straightforward, with acetonitrile and methanol being the most commonly used (and tetrahydrofuran, or THF, rarely). Gradient elution is generally preferred for complex multicomponent samples as, achieving the elution of all components with a single solvent strength within the desired retention factor (k) range of 1 to 10 under isocratic conditions can be challenging [19].
Selection of detector and wavelength:
After chromatographic separation, the analyte of interest is detected using suitable detectors [20]. Commonly used commercial detectors in liquid chromatography (LC) include UV detectors, fluorescence detectors, electrochemical detectors, refractive index (RI) detectors, and mass spectrometry (MS) detectors. The choice of detector depends on the sample characteristics and the analysis objectives. For multicomponent analysis, absorption spectra may shift to longer or shorter wavelengths compared to the compound. Therefore, it is essential to acquire and overlay the UV spectra of the target analyte and impurities, normalizing them to account for varying concentrations in the mixture. A wavelength should be selected to ensure an adequate response for most analytes [19,21].
Developing an analytic approach:
The initial step in developing an analytical method for RP-HPLC involves selecting key chromatographic parameters, including the mobile phase, column, mobile phase flow rate, and the pH of the mobile phse. These parameters are determined through a series of trials and are subsequently evaluated against system suitability criteria. Common system suitability parameters include a retention time exceeding 5 minutes, a theoretical plate count greater than 2000, a tailing factor below2, a resolution above 5, and a percent R.S.D. of analye peak areas in standard chromatograms not exceeding 2.0%. for the simultaneous estimation of two components, the detection wavelength is typically set at an isobestic point. Subsequently, the linearity of the drug is assessed to establish the concentration range over which it exhibits a linear relationship. A laboratory-prepared mixture is also tested to evaluate the practicality of the developed method for simultaneous estimation. Finally, the marketed formulation is analyzed by diluting it to fall within the established linear concentration range [22,23]
Sample preparation plays a crucial role in HPLC analysis, ensuring the solution is consistent and uniform for injection onto the column. The primary objective is to produce a sample aliquot that is free of significant interferences, does not harm the column, and is compatible with the chosen HPLC method. This compatibility requires the sample solvent to dissolve in the mobile phase without impacting retention or resolution. The process begins with sample collection and proceeds through to the injection of the sample onto the HPLC column [24].
Identify the method’s limitations and refine it using experimental design. Evaluate its performance under varying conditions, with different instrument configurations, and across diverse sample types [25].
Validation is the process of evaluating and providing objective evidence to confirm that specific requirements for a defined intended use are fulfilled. It involves assessing the method’s performance and demonstrating that it meets the established criteria [26].
Analytical Method Validation:
According to the international organization for Standardization (ISO), validation is the process of confirming that specified requirements are suitable for their intended purpose. Verification, in this context, is defined as providing objective evidence that a particular item meets the stated requirements [27]. Analytical method validation is the documented confirmation that a specific process consistently produces a product that satisfies its established criteria and quality standards. Validating an analytical method involves demonstrating through laboratory studies that the method’s performance characteristics meet the need of its intended analytical application. Any newly developed or modified method must undergo validation to ensure its effectiveness, confirming that it delivers reliable and consistent results regardless of the operator or the laboratory equipment used. The type of validation program required depends entirely on the specific method and its intended application. Method validation results are essential for assessing the quality, reliability, and consistency of analytical outcomes, forming a key part of good analytical practices. Properly functioning, well maintained, and calibrated equipment is crucial to the validation process. Analytical methods must undergo initial validation or revalidation as necessary [28]. It primarily involves five distinct steps, which are outlined as follows:
System quantification ensures that the instrument is suitable for the intended analysis, the materials are appropriate for making analytical decisions, and the anylysts possess the necessary training, skills and prior documentation. This includes comprehensive analytical methods, properly authorized protocols, and pre-established standards that have been thoroughly reviewed. If the overall quantifications of a system are neglected, identifying the root cause of any issues that arise will become significantly challenging [41].
Sampling helps in selecting a representative portion of the fabric, which is then subjected to evaluation. Choosing an appropriate sampling technique is crucial as it ensures that the selected sample accurately represents the entire material, enabling meaningful statistical inferences. The statistical literature provides extensive research on sampling methods; however, the associated costs and required for each method should be carefully considered in advance [41].
Sample preparation is a critical aspect of successful method validation. It is noted that sample preparation accounts for 60 to 80% of the work load and operational costs in an analytical laboratory. There is sample and well-documented literature on sample preparation. However, researchers must consider that the choice of a specific preparation method depends on factors such as analyte concentrations, the sample matrix, sample size, and the chosen instrumental technique [41].
Sample analysis involves utilizing instruments to obtain qualitative or quantitative data from samples with an acceptable level of uncertainty. The process can generally be understood as involving three interconnected components: input, converter, and output. The input (x) represents the concentration, while the output (y) denotes the response. The choice of a particular analytical method depends on various factors, such as the chemical properties of the analyte, the analyte concentration in the sample, the sample matrix, speed, cost, and other relevant considerations [41].
Data assessment aims to summarize and gain insights into a specific data set through the use of mathematical and statistical on methods. This process allows for the extraction of meaningful information and helps draw conclusions about the inputs, outputs, and, more importantly, the overall validation process [41].
Cleaning validation provides documented evidence with a high degree of assurance that a system or equipment can be consistently cleaned to meet predetermined specifications and criteria. It is a systematic process that demonstrates the effectiveness and reliability of cleaning pharmaceutical production equipment. The primary objective of cleaning validation is to confirm the capability of the cleaning process to effectively remove product residues, degradants, additives, excipients, or cleaning agents while controlling potential microbial contamination.
Reasons for cleaning validation
To avoid cross-contamination of pharmaceutical products and active pharmaceutical ingredients (API) with other products or microbes
Ensures compliance with regulatory requirements for maintaining cleanliness, safety, and quality in pharmaceutical manufacturing.
Ensures consistent internal control of manufacturing processes to maintain product quality.
Protects the quality and integrity of pharmaceutical products.
Facilitates the safe and effective reuse of manufacturing equipment [42,43].
Necessity of cleaning validation:
To ensure cleaning methods are effective and to eliminate risks associated with cross-contamination from APIs for detergents [42,43].
Cleaning validation Protocol:
A cleaning validation protocol typically includes:
Clearly defined goals of the validation
Roles and responsibilities for conducting and approving the validation study.
Defined interval between production completion and the start of cleaning procedures.
specific cleaning for each product, manufacturing device, or equipment.
Number of cleaning cycles to be performed consecutively.
processes for regular monitoring of equipment cleanliness.
methods for sample collection, including justification for the selected technique.
Specific areas for sampling and their rationale.
information on recovery studies, where applicable.
Methods used for analysis, including limits of detection (LOD) and quantification (LOQ).
Defined limits with reasoning for their selection [44]
Importance of validation:
The FDA, USP, and ICH recommend the following standard parameters.
The range of an analytical technique refers to the span between the highest and lowest analyte concentrations sample for which the method has demonstrated acceptable precision, accuracy, and linearity [29].
Linearity in a method refers to its capacity to generate test results that are directly proportional to the concentration of the analyte within a specified range. For HPLC methods, this linear relationship between detector response (such as peak area or height) and analyte concentration is assessed. The relationship can be directly established for the drug substance by either diluting a standard stock solution or independently weighing the drug substance. To demonstrate this, prepare the sample components using the proposed procedures. Linearity should be evaluated visually by examining a plot of signals against analyte concentration or content. When a linear relationship is observed, the results should be analyzed using suitable statistical techniques, such as regression analysis. The regression line can provide mathematical estimates to quantify the degree of linearity, often expressed as the variance around its slope. In certain cases, the analytical response may need to be characterized by the appropriate function of the analyte concentration. Commonly used linearity ranges and acceptance criteria are applied across various pharmaceutical methods [30].
In strategy approval, the items selectivity and specificity are often used interchangeably to describe the same concept. Specificity refers to the ability to distinctly identify and evaluate the analyte in the presence of other components that may need to be detected. The specificity of a testing system is assessed by comparing results from samples containing impurities, degradation products, or placebo components with results from samples that do not contain these interfering substances [31,32].
Accuracy refers to hoe close a measured value is to the true or accepted value. In practice, accuracy is determined by the difference between the mean measured value and the true value. This is typically assessed by applying the method to samples with known amounts of analyte, ensuring no interference, and comparing the results to standard and blank solutions. The accuracy is then expressed as a percentage of the analyte recovered in the assay based on the results, often referred to as the recovery of added known amounts of analyte [20].
Precision refers to the degree of consistency or scatter among a series of measurements obtained from multiple samplings of the same homogenous sample under specified conditions. It reflects the reproducibility of the analytical method and consists of two components: repeatability and intermediate precision.
Repeatability: pertains to the variability observed when a single analyst uses a single instrument. It does not differentiate between variations arising from the instrument or system and those from the sample preparation process. During method validation, repeatability is assessed by analyzing multiple replicates of a composite sample using the specified analytical method, and recovery values are calculated.
Intermediate precision: represents the variability within a laboratory, accounting for factors such as different days, instruments, or analysts [33].
The stability of standards and samples is assessed during validation under typical conditions, standard storage conditions, and occasionally within the instrument itself. This process helps determine whether any special storage requirements, such as refrigeration or light protection, are necessary [34].
The detection limit of a single analytical method is the minimum amount of analyte in a sample that can be detected, but not accurately quantified [35].
The quantitation limit of an individual analytical system is the smallest amount of analyte in a sample that can be measured with reliable accuracy and precision. This limit is a key parameter for quantifying low levels of substance in test samples and is especially useful for detecting contaminants or impurities [36].
The robustness of an analytical procedure refers to its ability to remain unaffected by small, intentional changes in method parameters, demonstrating its reliability and consistency under normal operating conditions [37].
Ruggedness of an analytical method refers to the consistency and reproducibility of test results when analyzing the same samples under varying conditions. These conditions may include differences in laboratories, analysts, instruments, reagent batches, assay times, temperatures, or days. It evaluates the method’s ability to deliver reliable results despite variations typically encountered between different laboratories or analysts [38].
Liquid chromatographic methods incorporate system suitability tests as a routine procedure to verify that the chromatographic system’s detection sensitivity, resolution, and reproducibility meet the required standards for analysis. These tests operate on the principle that the equipment, electronics, analytical procedures, and sample form an integrated system that can be assessed collectively. Key parameters such as peak resolution, theoretical plate count, peak tailing, and capacity are measured to evaluate the method’s suitability [39].
CONCLUSION:
In recent years, considerable focus has been placed on developing analytical techniques for drug identification, purity assessment, and quantification within the field of pharmaceutical analysis. This review offers a broad overview of HPLC method development and validation, presenting a straightforward approach to designing HPLC methods for compound separation. A thorough understanding of the primary compound’s physicochemical properties is essential before initiating HPLC method development. Additionally, the composition of the buffer and mobile phase, including its organic content and pH, plays a crucial role in influencing separation selectivity. Validation is an essential process in the pharmaceutical field ensuring that quality is integrated into the procedures supporting drug development and manufacturing. The optimized method is validated based on various parameters, such as range, linearity, specificity, accuracy, precision, limit of detection, robustness and system suitability, in accordance with ICH guidelines.
REFERENCES



Nikita Pabale*, Monali Khatke, Mansi Shelke, Vishweshwari Bhagat, Tanvi Kamble, A Review on HPLC Method Development and Validation, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 11, 1496-1509. https://doi.org/10.5281/zenodo.14234238
 10.5281/zenodo.14234238
10.5281/zenodo.14234238
