Department of Pharmaceutical Quality Assurance, Sandip Foundation’s, Sandip Institute of Pharmaceutical Sciences, Mahiravani, Dist.- Nashik, Maharashtra, India 422213.
The terms Sustained release used to identify drug delivery systems that are designed to achieve or extend therapeutic effect by continuously releasing medication over an extended period of time after administration of a single dose. There are several reasons for attractiveness of these dosage forms: provides increased bioavailability of drug product, reduction in the frequency of administration to prolong duration of effective blood levels, reduces the fluctuation of peak trough concentration and side effects and possibly improves the specific distribution of the drug. (1).
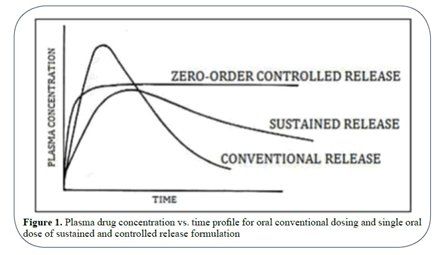
Fig. 1.1 Plasm drug concentration vs time profile
Advantages of Sustain Release Dosage Forms:
Disadvantages of Sustained Release Drug Delivery:
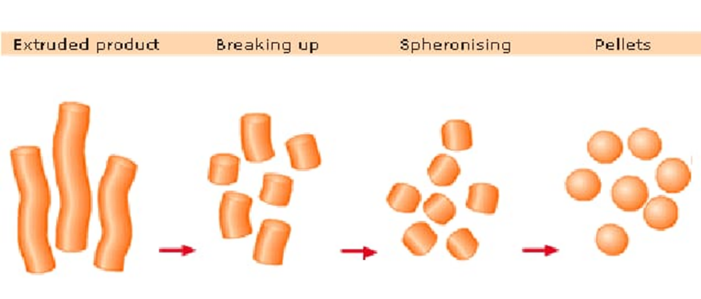
Extrusion and Spheronization technique :
The most promising method for the best administration of many strong medications with high systemic toxicity appears to be extrusion-spheronization. Due to the advantages of high loading capacity of active ingredient(s), limited size distribution, and cost effectiveness, it also provides enormous pharmaceutical applicability. These systems can also help with site-specific distribution upon application of a particular coat, improving the bioavailability of many medications. The focus of the current review is on the extrusion-spheronization process and the operational (extruder types, screen pressure, screw speed, temperature, moisture content, spheronization load, speed, and time) and formulation (excipients and drugs) variables that may have an impact on the final pellets' quality. The quality of the pellets is evaluated using a variety of techniques that take into account their size distribution, shape, friability, granule strength, density, porosity, flow characteristics, and surface roughness (4)

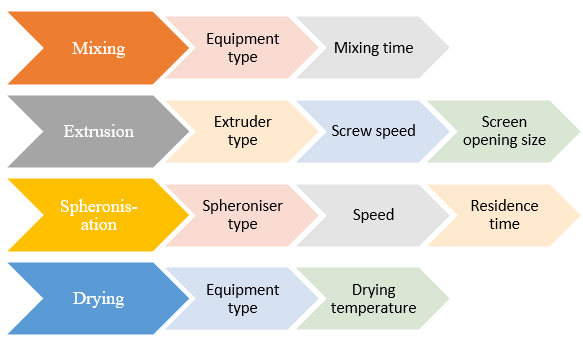
Fig. 1.2. Processing flowchart indicating individual process variables (18)
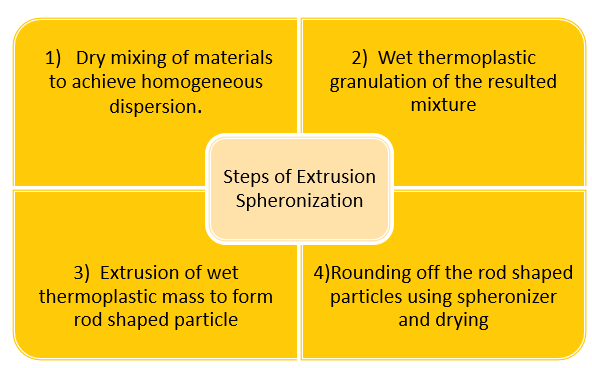
Formulation:
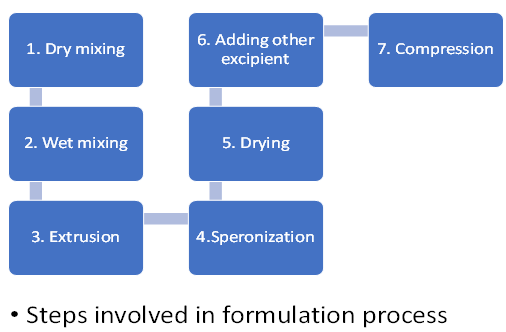
Formulation Procedure:
Wet granulation is a step in the formulation process for sustained released matrix tablets.
The process of wet granulation is frequently employed to create compressed tablets. The granules created using this method of grains on are more likely to meet all the physical requirements for tablet formulation since it is essentially a process of size enlargement comprising numerous phases and the use of an adhesive component known as binder.(5)
Procedure:
1.Forming a dump mass by weighing, grinding, and combining APIs with powder excipients. Water is used as the solvent.
2. Moving this wet mass from the cold melt extruder to modify the physical characteristics of the materials.
3. Screening the extruder material into pellets or granules with spheroids.(6)
Dry obtained granules with the help of hot air oven the temperature 600c for 20 minutes.
Literature Review:
|
Sr. No. |
Author |
Tittle |
Relevance to Present Work |
|
1. |
Batycky et al., Materials for direct compression. In: alderborn G, Nystrom C,eds. |
Pharmaceutical Powder Compaction Technology.New York,NY:Marcel.
|
Direct compression method |
|
2. |
Ballard,B.E., : Edited by J.R.Robinson.NewYork, Marcel Dekker (1978). |
An overview of prolonged action drug dosage forms |
sustained & controlled release drug delivery systems. |
|
3.
|
Brazel JG, Peck GH, Peppas YT., using neural formulations. J Control Release, 2000; 26(2): 211-215. |
Pharmaceutical granulation and tablet formulation.
|
Pharmaceutical granulation and tablet formulation network in sustained release. |
|
4. |
J.E.Hogan, Drug Dev. Ind. Pharm.15 (6,7), 975-999 (1989). |
Hydroxypropyl Methylcellulose sustained-Release Technology |
Information of HPMC |
|
5.
|
Sinha VR, Agrawal MK, Agarwal A, Singh G, Ghai D. ,Critical Reviews in Therapeutic Drug Carrier Systems. Begell House Inc.; (2009) |
Extrusion-spheronization: Process variables and characterization. |
Study of process variables. |
Objective Of The Study:
1. Salbutamol is used to treat the breathlessness, coughing, and wheezing that are signs of asthma and chronic obstructive pulmonary disease (COPD).
2.Sustained release Dosage forms for salbutamol sulphate are designed to release a medication at a set pace while keeping the drug level stable for a particular period of time.
5. The see-saw fluctuation in medication plasma levels is reduced by sustained release formulations.
6. The convenience of use and compliance of the patient are improved due to less frequent drug delivery.(8)
Formulation strategy for oral SRDDS:
1. Diffusion Sustained System
2. Dissolution Sustained System
3. Methods using ion exchange
4. Methods using osmotic pressure
5. pH independent formulation
6. Altered density formulation.(9)
Plan Of Work
Drug And Excipient Information:
5.1 Salbutamol Sulphate:
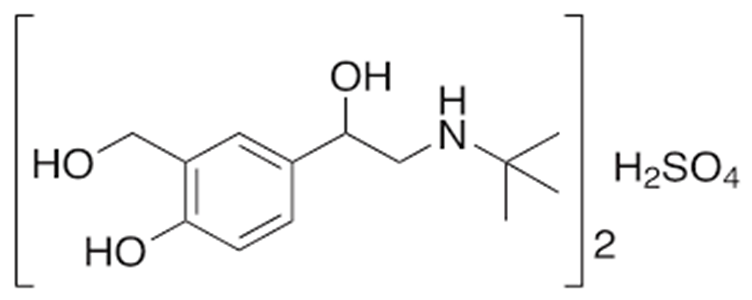
Fig.no.5.1 Salbutal sulphate chemical structure
Asthma and COPD are both treated with salbutamol, a short-acting, selective beta2-adrenergic receptor agonist. It has better selectivity for pulmonary beta receptors compared to beta1-adrenergic receptors found in the heart because it is 29 times more selective for beta2 receptors than beta1 receptors. As a racemic combination of the R- and S-isomers, salbutamol is created. The S-isomer has been linked to toxicity, while the R-isomer has a 150 times stronger affinity for beta2-receptors. Levalbuterol, the sole R-isomer of salbutamol, was created as a result. Levalbuterol is an enantiomerically pure form of salbutamol, but it is more expensive, preventing widespread use. Salbutamol is typically used to treat acute bronchospasm episodes brought on by bronchial asthma, chronic bronchitis, and other chronic bronchopulmonary diseases such chronic obstructive pulmonary disease (COPD). In order to prevent asthma brought on by exercise, it is also utilised.(10)
5.2hydroxypropylmethyl Cellulose (HPMC) :
The superior binder HPC exhibits good compactibility and similar binder efficiency whether added as a solution or in dry powder form. Water-soluble polymers made from cellulose called hydroxypropyl methylcellulose (HPMC) cellulose ethers are employed as essential binders in pharmaceutical operations. Since HPMC polymers function well with both soluble and insoluble pharmaceuticals as well as at high and low dose levels, they are regarded as adaptable binding agents.(11)
5.3 Lactose:
The diluent in this formulation is lactose. With the exception of lyophilized goods and newborn formulae, lactose is frequently employed as a filler or diluent in tablets and capsules. Additionally, lactose is employed in dry-powder inhalation as a diluent. Depending on the crystallisation and drying circumstances, lactose can take on a variety of isomeric forms, including lactose monohydrate, b-lactose anhydrous, and a-lactose anhydrous. A-lactose monohydrate, b-lactose anhydrous, and stable a-lactose anhydrous are the three stable crystalline forms of lactose. Lactose is a white to off-white powder or crystalline particle. Lactose has no smell and a mildly sweet flavour; a-lactose is about 20% sweeter than sucrose and b-lactose is 40% sweeter. [12]
5.4 Magnesium Stearate:
In this composition, the lubricant is magnesium stearate. Magnesium sterarate has the molecular formula C36H70MgO4. The molecular mass is 591.34. Magnesium stearate is a substance made of magnesium and a combination of solid organic acids, primarily magnesium stearate and magnesium palmitate (C32H62MgO4), in varying amounts. According to the PhEur 2005, magnesium stearate is a combination of magnesium salts of several fatty acids, primarily stearic acid and palmitic acid with trace amounts of other fatty acids. Magnesium stearate is a very fine, light white, precipitated or milled powder with a low bulk density, a slight stearic acid odour, and a distinctive flavour. When touched, the powder feels oily and immediately sticks to the skin. [13]
5.5 Talc:
Talc is employed in this composition as a glidant. A pure, hydrated magnesium silicate with a formula that resembles Mg6(Si2O5)4(OH)4 is talc. Iron and aluminium silicate may be present in trace levels. Talc is an extremely fine, crystalline, odourless, impalpable, unctuous, white to grayish-white powder. It is gentle to the touch, easily adheres to the skin, and doesn't feel gritty. Since talc is a stable substance, sterilisation can be accomplished by heating at 160°C for at least an hour. Talc needs to be kept dry and cool in a container that is tightly closed. [14]
5.6 Microcrystalline Cellulose (MCC):
MCC is employed as a disintegreat in this formulation. The nonproprietary designation for polyethylene oxide is USP-NF. Polyox, Polyoxiante, and Polyoxyethylene are synonyms. Polyethylene oxide is a chemical compound. According to the USP-NF, polyethylene oxide is an ethylene oxide nonionic homopolymer represented by the formula, where a represents the typical number of oxyethyl groups. A appropriate antioxidant or silicon diode up to 3% may be present.(15)
Materials And Instruments :(16)
Table. 6.1.List of equipment’s
|
Sr No |
Equipment |
Make |
|
1 |
Extruder |
VJ Instruments |
|
2 |
Dissolution Apparatus |
Shimatzu |
|
3 |
Fourier transform infrared (FTIR) spectrometer. |
Bruker optic Alpha II |
|
4 |
Spheronizer |
|
|
5 |
Weighing Machine |
WENSAR Instruments |
|
6 |
Ultraviolet Spectrophotometer |
Lab India Analytical |
|
7 |
Hardness Tester |
Monseto Tester |
|
8 |
Friability Apparatus |
Labline |
|
9 |
Disintegration Apparatus |
Electrolab |
|
10 |
Tablet Compression Machine |
RIMEK Karnawati |
Table. 6.2. List of excipients
|
Sr No |
Materials used |
Grade |
Role |
|
1 |
Salbutamol sulphate |
Industrial |
Bronchodilator |
|
2 |
Magnesium Stearate |
Laboratory |
Lubricant |
|
3 |
Talc |
Laboratory |
Glident |
|
4 |
Lactose |
Laboratory |
Diluent |
|
5 |
MCC |
Laboratory |
Disintegrant |
|
6 |
Hydroxypropylmethyl cellulose |
Laboratory |
Binder |
Experimental Work :
Preformulation:
Salbutamol is nearly insoluble in water, but it is soluble in organic solvents like methanol and chloroform.
The stock solution had a concentration of 0.1 mg/ml and was made in a 100 ml volumetric flask containing 10 mg of salbutamol and methanol. 0.2, 0.4, 0.6, 0.8, and 1 ml of the aforementioned solution were taken out and blended to make the volume in a 10 ml volumetric flask. Its concentration ranged from 2, 4, 6, 8, or 10 g/ml. By graphing the average absorbance values against the relevant drug concentration, the standard curve for salbutamol was produced.
Table 7.1. Solubility of salbutamol with methnol
|
Sr no. |
Concentration |
Absorbance |
|
1 |
0 |
0 |
|
2 |
2 |
0.177 |
|
3 |
4 |
0.346 |
|
4 |
6 |
0.532 |
|
5 |
8 |
0.725 |
|
6 |
10 |
0.935 |
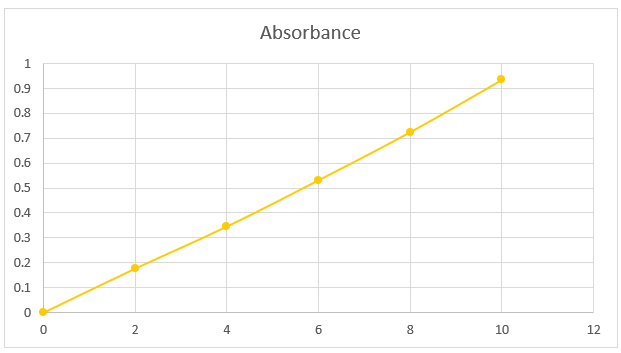
Fig. 7.1. Standard absorption curve of salbutamol sulphate
Standard curve of interaction with excipients :
The stock solution had a concentration of 0.1 mg/ml and was prepared in a 100 ml volumetric flask with 10 mg of salbutamol and HCL buffer (pH 6.8). 0.2, 0.4, 0.6, 0.8, and 1 ml of the aforementioned solution were taken out and blended to make the volume in a 10 ml volumetric flask. Its concentration ranged from 2, 4, 6, 8, or 10 g/ml. By graphing the average absorbance values against the relevant drug concentration, the standard curve for salbutamol was produced.
Table 7.2. Standard values of interaction with excipients
|
Sr no. |
Concentration |
Absorbance |
|
1 |
0 |
0 |
|
2 |
2 |
0.164 |
|
3 |
4 |
0.341 |
|
4 |
6 |
0.531 |
|
5 |
8 |
0.723 |
|
6 |
10 |
0.931 |
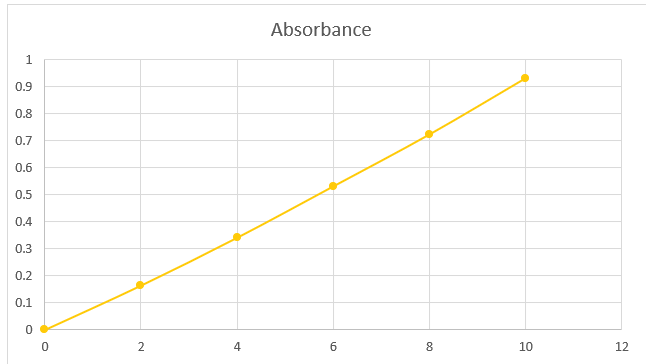
Fig 7.2. Standard curve of interaction with excipients
Table 7.3. Formula of salbutamol sulphate
|
Ingredients |
weight(mg/ml) |
|||
|
|
F1 |
F2 |
F3 |
F4 |
|
Salbutamol |
8 |
8 |
8 |
8 |
|
Lactose |
101.4 |
99.6 |
97.6 |
95.6 |
|
Microcystalline cellulose |
100 |
100 |
100 |
100 |
|
Talc |
2.2 |
2.2 |
2.2 |
2.2 |
|
Magnesium sterate |
2.2 |
2.2 |
2.2 |
2.2 |
|
Hydroxypropylmethyl cellulose |
36.2 |
38 |
40 |
42 |
Non-Official Tests:
1) General appearance
I) Organoleptic property
II) Size & Shape
2) Hardness
3) Friability Official tests
Official Tests:
1) Weight Variation
2) Drug Content
3) Dissolution
4) Disintegration
1.1 Non-Official Tests:
Table7.4 : Non-Official Tests Of Formulation
|
Type Of Test |
F1 |
F2 |
F3 |
F4 |
|
Size |
3mm |
3mm |
3mm |
3mm |
|
Shape |
Round |
Round |
Round |
Round |
|
Colour |
Off White |
Off White |
Off White |
Off White |
|
Odour |
Odourless |
Odourless |
Odourless |
Odourless |
|
Taste |
Bitter |
Bitter |
Bitter |
Bitter |
1.2 Hardness
The hardness of the tablet was measured using the Monsanto hardness tester. Tablet was placed between two anvils and a force (kg/cm2) was applied. The crushing strength that just caused the tablet to break was recorded. (17)

Fig. 7.3. Monsanto hardness tester
1.3 Friability:
The friability of the tablets was measured using the laboratory friability apparatus known as Roche friabilator. A pre-weighed sample of tablets was placed in the friabilator and operated for 100 revolutions at the rate of 25 rpm. These tablets were dedusted, reweighed and the percent friability was calculated using the following formulae:
F(%)= W1–W2 / W1 x 100
Where,
W1 = Initial weight of tablet
W2 = Final weight of tablet
The acceptance value for the tablets to pass friability was not more than 1% .(18)

Fig. 7.4. Roche friabilator
1.2 Official Tests:
Weight Variation:
From each batch 20 tablets were randomly selected and their average weight was calculated. The individual weight of each tablet was compared with the average weight of 20 tablets. The tablets were said to pass the weight variation test if they complied with the weight variation specifications as per I.P.(19)
Table 7.5. Standard of weight variation
|
USP Standard |
Max. % Difference |
BP/IP Standard |
|
130mg OR Less |
10% |
84mg OR Less |
|
130mg TO 324mg |
7.5% |
84mg TO 250mg |
|
More than 325 |
5% |
More than 250mg |

Fig 7.5 Weight balance
Drug Content:
7.5 Drug content and entrapment efficiency:
50 mg of the formulation was taken in 100 ml phosphate buffer solution. After filtration, 10 ml filtrate solution was taken in a volumetric flask and the volume was adjusted to 100 ml by phosphate buffer and the absorbance was taken at 276 nm. From the absorbance value, the amount of drug entrapped was determined using a standard curve.
The drug content in 50mg powder = 10.91mg
The drug content in 229.1mg powder= ‘x’ mg
Therefore x = 50 mg of the drug
The drug entrapment efficiency was calculated by using the following equation – (10)
Entrapment efficiency (%) = Calculated drug concentration/Theoretical drug concentration × 100 % (20)
iii) Dissolution test :
For 200 ml = 50 ml of PDP
For 4000 ml = x ml of PDP
x = 1000 ml of PDP
50 ml = 1.36 gm
1000 ml = x
x = 27.2 gm of PDP for 1000 ml of PDP solution.
for 100 ml = 0.85 ml of Conc HCL
for 1000 ml = 8.5 ml of Conc HCL
for 4000 ml = 34 ml of Conc HCL diluted with distilled water.
As mandated by USP, a dissolving study of the pellets was conducted. A phosphate buffer with a pH of 7.4 as well as in 0.1 N HCl. At a temperature of 37.5 °C and a paddle rotation speed of 50 rpm, the test was carried out using a USP dissolving apparatus type II. At each predefined time point, 5.0 mL of the dissolution media was removed and replaced with 5.0 mL of fresh medium. After that, Whatman filter paper was used to filter the dissolution samples. (21)
Table no.7.6. The dissolution profile of Salbutamol in phosphate buffer
|
Time |
Absorbance |
Concentration (ug/ml) |
Dilution Factor |
Concentration (mg/5ml) |
Concentration (mg/900ml) |
Cumulative core (mg/5ml) |
Cumulative core (ug/900ml) |
Cumulative core (mg/900ml) |
|
1 |
0.0104 |
0.1432 |
1.431 |
7.155 |
1287.9 |
7.155 |
1287.9 |
1.2879 |
|
2 |
0.0162 |
0.2538 |
2.538 |
12.69 |
2284.2 |
19.845 |
2291.355 |
2.2913 |
|
3 |
0.0192 |
0.3110 |
3.11 |
15.55 |
2799 |
35.395 |
2818.845 |
2.8188 |
|
4 |
0.0238 |
0.3988 |
3.988 |
19.94 |
3589.2 |
55.335 |
3624.595 |
3.6245 |
|
5 |
0.0283 |
0.4847 |
4.847 |
24.235 |
4362.3 |
79.57 |
4417.635 |
4,4176 |
|
6 |
0.0315 |
0.5458 |
5.458 |
27.29 |
4912.2 |
106.86 |
4991.77 |
4.9917 |
|
7 |
0.0342 |
0.5973 |
5.973 |
29.865 |
5375.7 |
136.725 |
5482.56 |
5.4825 |
|
8 |
0.0387 |
0.6832 |
6.832 |
34.16 |
6148.8 |
170.885 |
6285.525 |
6.2855 |
|
9 |
0.0441 |
0.7862 |
7.862 |
39.31 |
7075.8 |
210.195 |
7246.685 |
7.2466 |
|
10 |
0.0455 |
0.8129 |
8.129 |
40.645 |
7316.1 |
250.84 |
7526.295 |
7.5262 |
|
11 |
0.0494 |
0.8874 |
8.874 |
44.37 |
7986.6 |
295.21 |
8237.44 |
8.2374 |
|
12 |
0.0514 |
0.9255 |
9.255 |
46.275 |
8329.5 |
341.485 |
8624.71 |
8.6247 |
Table no. 7.7. Percentage of drug release of Salbutamol:
|
Time (in hr) |
% Drug Release |
|
1 |
14.80 % |
|
2 |
26.33 % |
|
3 |
32.4 % |
|
4 |
41.66 % |
|
5 |
50.77 % |
|
6 |
57.37 % |
|
7 |
63.01 % |
|
8 |
72.24 % |
|
9 |
83.29 % |
|
10 |
86.50 % |
|
11 |
94.68 % |
|
12 |
99.10 % |
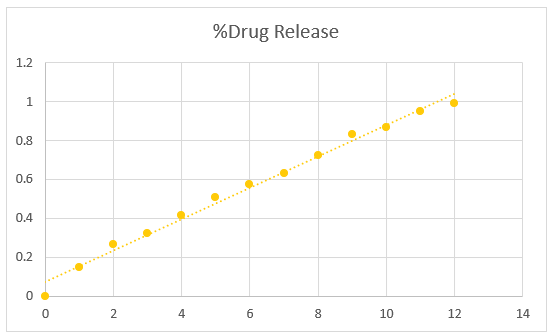
Fig 7.6. Graph of percentage drug release of salbutamol
Table no. 7.8. Percentage drug release of Salbutamol sulphate in .01N HCL
|
Time (min) |
%Drug Release |
|||
|
|
F1 |
F2 |
F3 |
F4 |
|
0 |
0 |
0 |
0 |
0 |
|
1 |
14.80% |
10.50 |
16.60 |
10.20 |
|
2 |
26.33% |
22.50 |
29.50 |
18.35 |
|
3 |
32.40 |
29.20 |
36.50 |
27.56 |
|
4 |
41.66 |
39.70 |
45.80 |
36.54 |
|
5 |
50.77 |
47.20 |
54.90 |
45.21 |
|
6 |
57.37 |
53.60 |
59.30 |
50.65 |
|
7 |
63.01 |
59.50 |
66.10 |
57.25 |
|
8 |
72.24 |
69.30 |
75.30 |
66.53 |
|
9 |
83.29 |
80.40 |
86.54 |
76.24 |
|
10 |
86.50 |
82.80 |
88.21 |
79.64 |
|
11 |
94.68 |
91.62 |
95.41 |
88.78 |
|
12 |
99.10 |
95.20 |
97.90 |
92.56 |
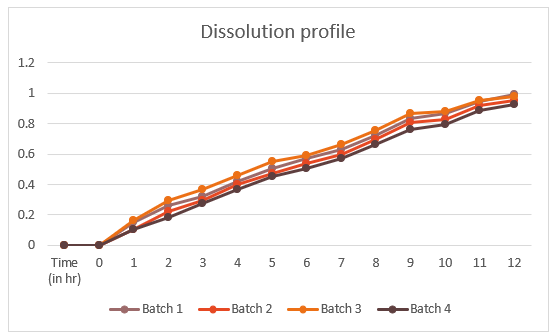
Fig. 7.7 Graph of dissolution profile

Fig 7.8 Dissolution apparatus
Disintegration test :
The disintegration test is used to demonstrate how quickly a tablet disintegrates into tiny pieces, increasing the surface area and medication availability when ingested by a patient. We use a basket that can accommodate one to six tablets to conduct a disintegration test on tablets. This is then elevated and lowered into a water-filled beaker to mimic the 37°C environment of the stomach. Perforated plastic discs are placed on top of floating tablets or capsules to keep them submerged in water. When there is no longer any debris in the mesh, the tablet disintegration time is measured.(22)
RESULT:
Size, shape, colour, and other physical characteristics like hardness and friability were all within acceptable ranges. Compressed tablets met all physical specifications set by the government.
|
Sr no. |
Parameters |
F1 |
F2 |
F3 |
F4 |
|
1 |
Size : |
3mm |
3.1mm |
3mm |
3mm |
|
2 |
Shape : |
Round |
Round |
Round |
Round |
|
3 |
Colour : |
Off white |
Off white |
Off white |
Off white |
|
4 |
Hardness (kg/cm2) |
5 |
4.5 |
5.45 |
4 |
|
5 |
Friability : |
0.77% |
0.84% |
0.60% |
0.72% |
|
6 |
Weight variation |
250 mg tablet (IP limit (+ / -) 7.5%) 268mg to 252 mg |
|||
|
7 |
Dissolution test |
Drug release for over 12 hrs with 99.10 % drug releases irrespective of excipient content. |
|||
|
8 |
Disintegration time (hr) |
3 hr 15 mins |
2 hr 20 mins |
2 hr 30mins |
2 hr 40 mins |
DISCUSSION:
Sustained release matrix tablets are the subject of the current article. The idea of matrix tablets might easily and effectively accomplish the continuous release goal. Matrix tablets offer better patient compliance than their conventional counterparts, maintain consistent plasma drug concentration levels, reduce the risk of toxicity, and once-daily prescription therapy lowers overall treatment expenses. It is advantageous to reduce unreasonable drug use (especially of antibiotics) and to manage chronic illnesses by keeping medication concentrations within therapeutic ranges. Additionally, it is an economical tactic.
CONCLUSION:
It is discovered that compared to their rivals, oral sustained release pills administer the medication differently. It is a productive method for deciding on therapy objectives while assuring maximum patient compliance. But numerous physicochemical parameters need to be adjusted precisely. The matrix pill is helpful in resolving the problems with conventional dosage forms. In addition to the many benefits associated with it, its cost effectiveness and once-daily dose are the main benefits. It might easily lead the market by displacing its rivals due to its key features and increased patient compliance.
REFERENCES

Shounak Mande*, A Research Article on Sustained Release Tablet of Salbutamol Sulphate, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 1, 1429-1441. https://doi.org/10.5281/zenodo.14684858
 10.5281/zenodo.14684858
10.5281/zenodo.14684858
