Dr. K. V. Subba Reddy Institute of Pharmacy.
Tay-Sachs disease is a genetic disorder impacting the central nervous system due to HEXA enzyme deficiency, resulting in GM2 ganglioside accumulation and progressive neurological decline. Initially documented by Waren Tay and Bernard Sachs in Ashkenazi Jewish populations, the disease exhibits three types: infantile, juvenile, and late-onset, each differing in symptom onset and progression. Current therapies are primarily supportive, focusing on symptom management, with ongoing research exploring various treatment strategies. Enzyme Replacement Therapy (ERT) aims to replace the non-functional HEXA enzyme, though challenges exist in crossing the blood-brain barrier. Substrate Reduction Therapy (SRT) seeks to inhibit the synthesis of GM2 gangliosides, with compounds like miglustat showing potential but limited clinical success. Gene therapy, which introduces a functional HEXA gene, has shown promise in animal studies, while pharmacological chaperones aim to stabilize misfolded enzymes and increase their activity. Despite the constraints of current treatments, research developments in these areas offer hope for improving outcomes for individuals affected by Tay-Sachs disease, emphasizing the need for continued investigation to enhance therapy efficacy and patient quality of life. Advances in alternative strategies, such as gene therapy and pharmacological agents, could play a critical role in managing this rare condition.
Tay-Sachs disease is a genetic disorder that affects the central nervous system by disrupting the breakdown of GM2 gangliosides due to a deficiency in the HEX-A enzyme. This leads to the accumulation of fatty substances in nerve cells, causing them to malfunction and die, which results in progressive neurological symptoms.1 Waren Tay and Bernard Sachs were physicians who first described Tay-Sachs disease, noting its progression and distinguishing it from other neurological disorders. Tay reported cases among Ashkenazi Jewish families in 1881, while Sachs later shared similar findings. The disease is characterized by progressive mental and physical decline, with no known preventive measures or effective treatments. In 1969, researchers identified the enzyme defect causing Tay-Sachs and developed diagnostic assays to detect it. This led to protocols for newborn testing, carrier screening, and prenatal diagnosis by the early 1970s. By 1979, three variant forms of GM2 gangliosidosis were identified, improving carrier testing accuracy.2
Tay-Sachs is classified into three types based on age of onset:
1. Infantile Tay-Sachs: The most severe form, noticeable by six months, with symptoms like loss of motor skills, increased startle reactions, and vision and hearing loss, leading to significant neurological impairment.
2. Juvenile Tay-Sachs: Appears between ages 2 and 5, characterized by muscle weakness, seizures, and loss of mobility, progressing more slowly but still life-limiting.
3. Late-onset Tay-Sachs (LOTS): Manifests in adolescence or adulthood, with symptoms such as muscle weakness, speech difficulties, and psychiatric issues, progressing much more slowly than the earlier forms, allowing for a longer lifespan.1 Tay-Sachs disease is rare, occurring in approximately 1 in 100,000 live births in the general U.S. population, with a carrier frequency of about 1 in 250. According to the epidemiological studies Tay Sachs disease is more commonly seen in the Ashkenazi Jewish descent where studies indicate the prevalence of 1 in 29 and an incidence of 1 in 3,500 live births affected by the disease.3 Tay-Sachs disease is caused by beta-hexosaminidase A (Hex A) deficiency, responsible for GM2 ganglioside degradation. Alpha and beta subunits of Hex A are synthesized at the endoplasmic reticulum.
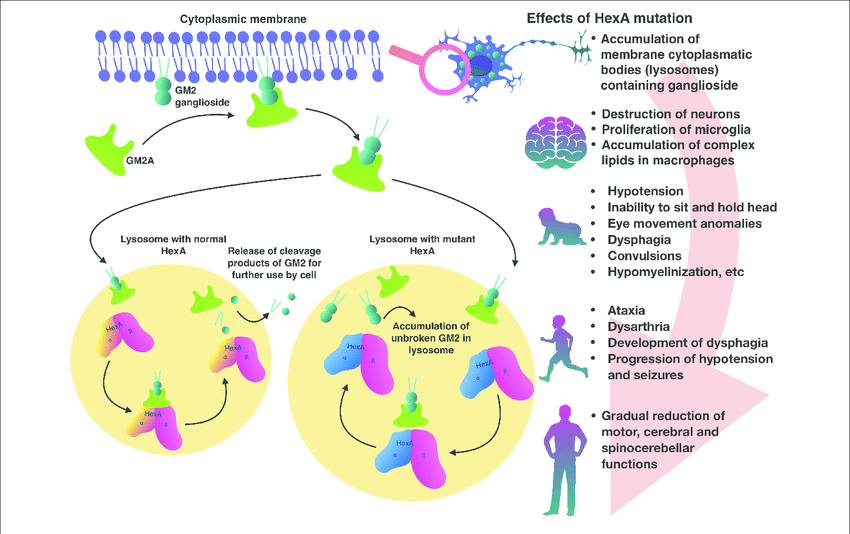
Fig. 1: Pathogenesis of Tay Sachs Disease4
Symptoms of infantile Tay-Sachs disease typically begin between 3 to 6 months of age and include:
Tay-Sachs disease should be suspected when initial symptoms appear, such as excessive responses to sound and a progressive loss of previously learned motor skills, leading to psychomotor retardation. Testing for hexosaminidase A activity can confirm the diagnosis, with low or absent levels (normal range: >50.0%) indicating the disease. Additionally, molecular genetic studies can identify genetic variations in the HexA gene. The form of Tay-Sachs—whether infantile, juvenile, or late-onset—depends on the age of onset, disease progression, and age at death. Biomarkers associated with gangliosidoses can help identify the disease and its distinct neuropathology, as their abnormal elevation correlates with the severity of clinical symptoms and the absence of other neurodegenerative lysosomal disorders.4 Treatment for Tay-Sachs disease is primarily supportive, focusing on providing adequate nutrition, controlling seizures, managing infections, protecting the airway, and implementing early physical and occupational therapy. Seizure management often requires multiple antiepileptic medications, with frequent adjustments needed as seizure patterns change.
1. Enzyme Replacement Therapy: While promising, this therapy faces challenges due to the inability of the enzyme to cross the blood-brain barrier. Recent studies using a recombinant chimeric protein that facilitates entry into the brain have shown encouraging results.
2. Enzyme Enhancing Therapy: This approach aims to stabilize the enzyme using pharmacological chaperones, which are enzyme-specific competitive inhibitors. A trial with the Hex A inhibitor pyrimethamine showed increased Hex A levels, but clinical benefits remain unreported.
3. Substrate Reduction Therapy: This therapy seeks to balance substrate synthesis with reduced enzyme activity. The drug miglustat has shown success in animal models but has not been approved for human use by the FDA.
4. Gene Therapy: Given that Tay-Sachs is caused by a single genetic disorder, gene therapy is a promising option. Progress has been made with adeno-associated virus-based vectors, although limitations exist regarding their capacity to carry the necessary constructs.
5. Bone Marrow Transplantation: This approach involves transplanting modified neural cells that express the HEXA gene.1
Objectives
MATERIALS AND METHODS
This is a review which includes the information about the treatment strategies to manage Tay Sachs disease (TSD) and its effectiveness. The peer reviewed articles from about 20 years were considered. Around 12 articles were selected using which various treatment options were analysed and discussed. The materials by Akeboshi7, Smith6, Kodoma9 were used to know about the advances in Enzyme Replacement Therapy. The articles by Leal11, Solovyeva10 were used to understand how Substrate Reduction Therapy is used in the management of Tay Sachs Disease. From few research articles the models through which gene therapy can be useful in managing Tay Sachs Disease was studied. Likely the other therapeutic strategies like HexA enzyme activity and Pharmacological Chaperones were studied by using articles by Smith6 and Maegawa15, Osher16 respectively.
DISCUSSION
Enzyme Replacement Therapy (ERT) for Tay-Sachs Disease:
Enzyme Replacement Therapy (ERT) has emerged as a potential treatment for Tay-Sachs disease (TSD) and other lysosomal storage disorders. The primary goal of ERT is to provide a functional replacement for the HEXA enzyme, which is non-functional in individuals affected by TSD. However, the efficacy of this approach is significantly hindered by the enzyme's large size, which prevents it from effectively crossing the blood-brain barrier (BBB) — a critical barrier for delivering therapeutic agents to the central nervous system (CNS). (Picache JA et al., 2022)5
Challenges of ERT in TSD
The inability of the HEXA enzyme to traverse the BBB poses a significant challenge for traditional intravenous administration methods. Current research has explored alternative strategies, including the direct administration of the HEXA enzyme into the cerebrospinal fluid (CSF).
Animal Models in Research
To further investigate TSD, researchers have utilized Jacob sheep as a model organism. This rare breed is particularly valuable due to its susceptibility to TSD, with pathophysiological characteristics and gene mutations closely mirroring those found in humans. Notably, a specific mutation in the HEXA complementary deoxyribonucleic acid (cDNA) of these sheep, known as G444R, has been identified as a missense mutation. This mutation results from a nucleotide change at exon 11, which disrupts the splicing process before transcription can occur. (Smith et al., 2021)6
Advancements in ERT Development
For ERT to be effective in treating TSD, it must include both functional alpha and beta subunits of the HexA enzyme. Several research groups have successfully developed functional HexA enzymes that have shown promise in alleviating disease phenotypes in vitro (Akeboshi et al., 2007; Tsuji et al., 2011)7,8. However, the challenge remains that traditional intravenous administration has not effectively addressed CNS symptoms due to the enzyme's size. To overcome the limitations of BBB permeability, researchers have explored alternative routes for HexA administration. One promising approach involves intracerebroventricular (ICV) injections of a modified HexB enzyme, which possesses both HexA and HexB activities (Matsuoka et al., 2011).9
While ERT presents a hopeful avenue for treating Tay-Sachs disease, significant challenges remain, particularly regarding the delivery of the HEXA enzyme to the CNS. Ongoing research into alternative administration routes and the use of animal models like Jacob sheep will be crucial in advancing our understanding and treatment of this devastating condition. Continued exploration of innovative strategies may ultimately lead to effective therapies that can improve the quality of life for individuals affected by TSD (Smith et al., 2021)6.
Substrate Reduction Therapy (SRT) for Tay-Sachs Disease:
Substrate Reduction Therapy (SRT) is an innovative treatment strategy aimed at managing Tay-Sachs disease (TSD) by utilizing enzymes to enhance the breakdown of GM2 gangliosides in the central nervous system (CNS). This approach seeks to compensate for the absence of the HEXA enzyme, thereby preventing the accumulation of lipids that contribute to the disease's symptoms and progression (Smith et al., 2021)6.
Mechanism of Action
The primary goal of SRT is to inhibit the production of the accumulating substrate, specifically GM2 gangliosides, in individuals with TSD. One enzyme that has garnered attention in this context is sialidase. By facilitating the metabolism of GM2 gangliosides, sialidase could potentially bypass the genetic defect associated with TSD, promoting healthier lipid metabolism. However, despite its promise, sialidase has not yet been developed into a safe and effective pharmacological treatment.
Miglustat: A Potential SRT Agent
Miglustat is another compound under investigation for its potential role in treating TSD. It functions as a competitive inhibitor of the glucosylceramide synthase enzyme, which is crucial for the synthesis of GM2 gangliosides. By inhibiting this enzyme, miglustat aims to reduce the levels of GM2 gangliosides in the CNS. In preclinical studies, miglustat demonstrated partial efficacy in animal models, successfully reducing GM2 levels in the brains of TSD mouse models by approximately 50% (Solovyeva et al., 2018)10. However, clinical trials have yielded mixed results. Although miglustat was tested in two infantile patients, it failed to prevent the progression of neurological symptoms (Leal et al., 2020).11 Further assessments included five patients with the juvenile form of GM2 gangliosidosis over a two-year period, which also showed no improvement in neurological signs and symptoms (B. Shapiro et al., 2009)12. Implementing SRT, including miglustat, during the early stages of disease progression may provide a better opportunity to prevent or mitigate neurodegenerative signs and symptoms. Substrate Reduction Therapy represents a promising avenue for the treatment of Tay-Sachs disease, with ongoing research into various enzymatic approaches, including sialidase and miglustat.
Gene Therapy for Tay-Sachs Disease
In recent years, gene therapy has emerged as one of the most significant advancements in the treatment of Tay-Sachs disease (TSD). This innovative approach focuses on introducing a functional HEXA gene to restore the production of hexosaminidase A, the enzyme that is deficient in individuals with TSD.
Mechanism of Gene Therapy
The primary goal of gene therapy for Tay-Sachs disease is to correct the genetic mutation responsible for the absence of the HEXA enzyme. By delivering a functional copy of the HEXA gene into the patient's cells, scientists aim to enable the production of hexosaminidase A, thereby preventing the accumulation of GM2 gangliosides that leads to the neurological symptoms associated with TSD.13
Advancements in Research
Early trials in animal models have shown promising results, with significant improvements in neurological function observed following the introduction of the functional HEXA gene. These findings provide a strong foundation for further research and development in this area. Researchers led by Martino conducted a groundbreaking experiment on gene therapy for Tay-Sachs disease by using a non-replicating herpes simplex viral vector (HSV-1) to restore the missing HexA gene in mice affected by the condition. Tay-Sachs is caused by a deficiency in the HexA enzyme, resulting in harmful GM2 ganglioside accumulation in nerve cells. The experiment involved dividing the mice into five groups, three of which received varying doses of the HSV-1 vector directly injected into the internal capsule of the brain, a region crucial for motor function. The results were promising: the mice that received the highest dose showed a complete clearance of GM2 gangliosides, while those with lower doses experienced partial improvement. Notably, the restoration of enzyme activity was observed throughout the entire central nervous system, indicating effective diffusion from the injection site, which contributed to mitigating the symptoms of Tay-Sachs. Overall, the study showcased the potential of using HSV-1 as a method for delivering therapeutic genes, paving the way for innovative treatments for genetic disorders like Tay-Sachs in the future.14
Role of Pharmacological Chaperones in Tay sachs disease
In Tay-Sachs disease (TSD), mutations can result in misfolded HexA enzymes that are quickly degraded before reaching the lysosome, where they are needed to break down GM2 gangliosides.
Pharmacological chaperones can promote proper folding of enzymes. One such compound, pyrimethamine (PYR), has been shown to inhibit HexA degradation long enough for the enzyme to complete its journey to the lysosome and sustain GM2 hydrolysis in vitro (Maegawa et al., 2007)15. PYR binds to domain II of HexA, stabilizing the enzyme in cells with the late-onset mutation alphaG269S.
Clinical Trials and Limitations
While PYR is approved for treating toxoplasmosis and has been tested in TSD clinical trials, the outcomes were limited. Patients experienced only temporary increases in HEXA activity without significant symptom improvement, alongside undesirable side effects (Osher et al., 2015).16
New Developments in Chaperone Compounds
Recent research has shifted toward small molecule binders that act as chaperones without inhibitory effects. These new compounds aim to enhance the functionality of mutant enzymes more effectively than traditional inhibitor chaperones, potentially offering better therapeutic options for TSD patients (Liguori et al., 2020)17. Here’s a brief summary of the alternative approach for treating Tay-Sachs disease, with side headings:
Increasing HEXA Enzyme Activity to manage Tay sachs disease
One investigated strategy for treating Tay-Sachs disease involves enhancing the activity of the HEXA enzyme to mitigate the effects of the enzyme deficiency characteristic of the disease. This approach is mainly targeted at patients with late-onset Tay-Sachs disease, as those with infantile Tay-Sachs typically lack the enzyme completely and would not benefit from increased activity.
Role of Pyrimethamine
Pyrimethamine is a drug that has shown some potential in increasing HEXA enzyme activity. However, its overall efficacy remains limited, as the activity of β-hexosaminidase A in treated patients is still significantly lower than in unaffected individuals. This suggests that while there may be some therapeutic effect, it is not sufficient to entirely overcome the enzyme deficiency associated with Tay-Sachs disease. (Smith et al., 2021)6.
CONCLUSION
Tay-Sachs disease is a severe genetic disorder caused by HEXA enzyme deficiency, leading to GM2 ganglioside accumulation and progressive neurological decline, particularly among Ashkenazi Jews. While current treatments focus on supportive care, ongoing research into innovative therapies, including enzyme replacement, substrate reduction, gene therapy, and pharmacological chaperones, holds promise for more effective management. Despite challenges, advancements in these treatment modalities may improve outcomes and quality of life for affected individuals, warranting continued exploration and clinical trials.
REFRENCES



Dr. M. Govardhan*, Alefiya Kotawala, Shaik Rizwana, A Comprehensive Review on Treatment Strategies of Tay Sachs Disease, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 3, 775-782. https://doi.org/10.5281/zenodo.14999822
 10.5281/zenodo.14999822
10.5281/zenodo.14999822
