1Sun Pharmaceutical Industries Limited, India
2School of Pharmaceutical Education and Research (SPER) Jamia Hamdard (Hamdard University)
3Amity Institute of Pharmacy, Amity University, Noida
Tacrolimus is a commonly prescribed immunosuppressant medication for prevention of organ transplant rejection. After organ transplantation, most of the patients are discharged and prescribed with tacrolimus medication due its high immunosuppressive efficacy. Although, tacrolimus is a go-to medication for the organ transplantation, it comes with numerous challenges such as narrow therapeutic index, high intra and interpatient variability in pharmacokinetics and the associated adverse effects. Therefore, it creates major difficulties for the clinicians by making the regular therapeutic drug monitoring and dose adjustment necessary. This review highlights various aspects including pharmacokinetics, food and drug interactions and genetics which are critical to understand the risk factors associated with tacrolimus and the measures required to overcome them. In particular, the information on human pharmacokinetics of tacrolimus would assist in further dose optimization with the defined pharmacokinetic-pharmacodynamic relationship.
A significant rise in organ transplants has been observed over the last 10 years with good success rate due to advancement in the surgical procedures and the availability of post-transplant medications. The drugs prescribed after an organ transplant play a pivotal role in the success of the overall procedure. One of the drug categories used are immunosuppressants, which prevent the newly transplanted organ from being attacked by the immune system of recipient. There are different categories of immunosuppressants that are reported according to their pharmacological background, such as, calcineurin (specific T-cell) inhibitors, antiproliferative (cytotoxic) drugs, glucocorticoids and antibodies. Tacrolimus is one such drug that belong to calcineurin category and is being widely used. It has shown comparatively better safety profile and extended survival of the patients. Despite being a go-to drug, it has some limitation, namely, lower bioavailability, pharmacokinetic variability and toxicity issues [1]
Tacrolimus or Fujimycin or FK-506, is a macrolide lactone (figure 1) derived from Streptomyces tsukubaensis, a type of soil bacterium. It is structurally related to macrolide antibiotics like erythromycin but functions as an immunosuppressant rather than an antibiotic.
It has a molecular weight of 804.0 g/mol and an XLogP3 value of 2.7, indicating moderate lipophilicity. It possesses three hydrogen bond donors, twelve hydrogen bond acceptors, and seven rotatable bonds, contributing to its structural flexibility. With an exact and monoisotopic mass of 803.48197664 Da, tacrolimus exhibits a high complexity of 1480 and a topological polar surface area of 178 ?². The compound, which has 57 heavy atoms and 14 defined stereocenters, is stable under recommended storage conditions. It forms colourless prisms from acetonitrile, has a melting point of 126°C, and is insoluble in water but soluble in organic solvents such as methanol, ethanol, and chloroform. Additionally, it has a specific optical rotation of -84.4° at 23°C and displays a collision cross-section of approximately 267–277 ?², depending on the ionization method used.

Figure 1: Chemical structure of Tacrolimus
Kurakado et al. [3] demonstrated that tacrolimus inhibits stress responses and hyphal formation in Trichosporon asahii via the calcineurin signalling pathway, suggesting antifungal potential. Antypenko et al. [4] performed an in silico molecular docking study showing tacrolimus, when combined with cyproconazole and other antifungals, enhances fungal chitin deacetylase inhibition. Fagone et al. [5] identified altered molecular pathways in Alzheimer's disease and suggested tacrolimus as a potential repurposed drug through in silico modelling. Walker et al. [6] explored drug repurposing for status epilepticus, indicating tacrolimus' possible neuroprotective and anti-seizure properties. Adiwidjaja et al. [7] applied physiologically based pharmacokinetic modelling to predict tacrolimus metabolism concerns when taken with Schisandra sphenanthera. Mofed et al. [8] examined in silico and in vitro effects of mTOR inhibitors, including tacrolimus analogs, in inducing apoptosis in hepatocellular carcinoma cells and inhibiting hepatitis C virus. Zhang et al. [9] investigated the circFASN/miR-33a pathway and its role in tacrolimus-induced hepatic triglyceride dysregulation, revealing metabolic side effects. Karwasra et al. [10] studied the impact of pomegranate rind extract on tacrolimus pharmacokinetics, showing significant drug concentration fluctuations due to food interactions. Mevizou et al. [11] revisited tacrolimus metabolite pharmacodynamics using computational methods, providing insights into its metabolic pathways. Gobeil et al. [12] designed FK506 analogs through in silico structural modeling, developing calcineurin inhibitors with reduced immunosuppressive effects. Faelens et al. [13] created an in-silico model-informed precision dosing framework for tacrolimus in kidney transplant recipients to enhance individualized therapy. Chen et al. [14] explored the FKBP1A/SLC3A2 axis using in silico analysis, highlighting tacrolimus' potential immunomodulatory effects in breast cancer. Vittorio et al. [15] used in silico strategies to target mTOR at the rapamycin binding site, providing insights for immunosuppressive drug design. Shang et al. [16] conducted an in-silico study on FKBP5 blockade, linking it to stress-associated disorders and tacrolimus-related pathways.
MECHANISM OF ACTION
Tacrolimus penetrates across the cellular membrane inside T-cells, reaching the cytoplasm, where it interacts with the immunophilin FKBP12 (FK506-associated protein-12KDa), while cyclosporine A attaches to cyclophilins. These binding interactions suppress the function of calcineurin [also recognized as PP2B (protein phosphatase-2B)], a calcium- and calmodulin-influenced serine/threonine phosphatase, obstructing its approach to substrates like NFAT (nuclear factors of activated T-cells) group members (NFAT1, NFAT2, and NFAT4) and their dephosphorylation, which is vital for controlling cellular messaging. Once the cytoplasmic percentage of NFAT undergoes dephosphorylation, it migrates toward the nucleus, forming an engaged transcription factor complex that regulates genes linked to B-cell activation, including interleukin-4 (IL-4) and CD40 ligand, as well as genes impacting T-cell multiplication and differentiation, such as interleukins 2 (IL-2), 3 (IL-3), 4 (IL-4), and 5 (IL-5). Thus, cytoplasmic absorbed tacrolimus disrupts calcineurin pathways, reducing IL-2 synthesis. Additionally, calcineurin enables a secondary surge of IL-2 expression through the transcription factor NF-κB, where calcineurin causes IκB breakdown, permitting NF-κB-guided expression of inflammatory-related genes. Consequently, suppression of calcineurin through tacrolimus raises the portion of NF-κB attached to IκB, thereby halting NF-κB-mediated gene transcription of inflammatory elements (figure 2).
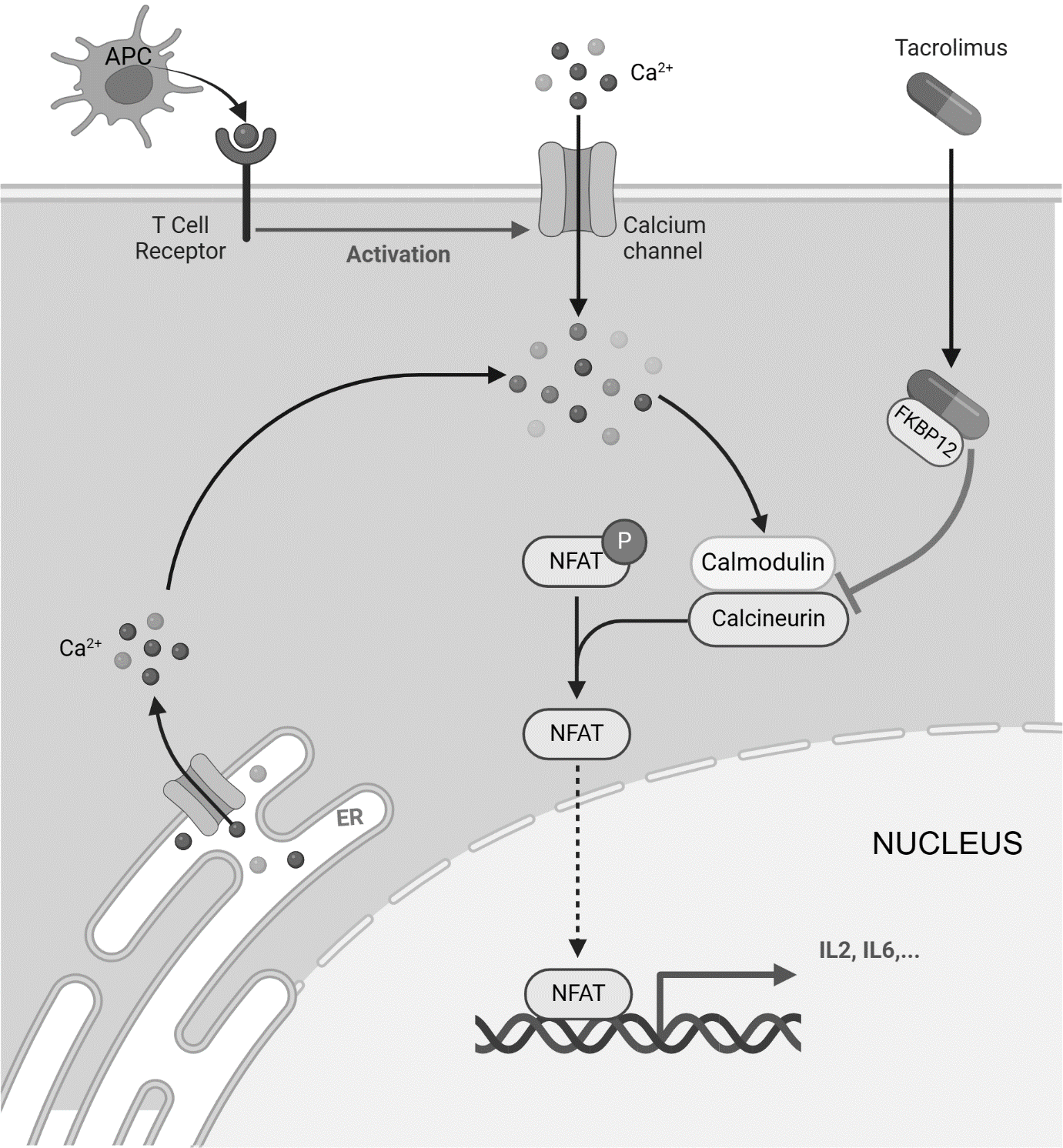
Figure 2: Schematic diagrammatic representation of action mechanism of tacrolimus. Tacrolimus binds to FKBP12, forming a complex that inhibits calcineurin, preventing NFAT dephosphorylation and its translocation to the nucleus. This blocks the transcription of IL-2 and other pro-inflammatory cytokines, suppressing T-cell activation and immune response
Pharmacodynamic studies on tacrolimus have provided critical insights into its therapeutic monitoring, dosing strategies, efficacy, and safety across a range of clinical conditions. Xiao et al. (2024) [18] identified critical values for therapeutic drug monitoring, while Han et al. (2024) [19] and Martischang et al. (2024) [20] further delineated optimum trough levels and steady-state concentrations, respectively, essential for graft survival and effective immunosuppression. Machine learning and physiologically based pharmacokinetic (PBPK) modelling approaches have emerged as promising tools to optimize dosing, as shown by Yoon et al. (2024) [21] in liver transplant recipients and Xu et al. (2025) [22] in pregnant women. Similarly, Guan et al. (2024) [23] applied PBPK modelling to understand CYP3A4/3A5 maturation and its effects on paediatric HSCT patients. The impact of pharmacogenomics was highlighted by Hannachi et al. (2024) [24] and Mohamed et al. (2024) [25], who analyzed the role of CYP3A polymorphisms and genetic variants in African American populations, while Zhang et al. (2024) [26] examined the influence of GSTM3 rs7483 in liver transplant patients. Drug–drug interactions were examined by Li et al. (2024) [27], who found that Paxlovid significantly alters tacrolimus concentrations. In renal transplantation, Chamoun et al. (2024) [28] demonstrated that transcriptomic markers of rejection correlate with tacrolimus exposure and graft outcomes, while Maslauskiene et al. (2024) [29] emphasized the importance of tacrolimus metabolism in predicting long-term success. Takeuchi et al. (2024) [30] and van Schaik et al. (2024) [31] provided long-term safety and efficacy data for lupus nephritis patients, reinforcing the drug’s role in autoimmune nephropathies. In gastrointestinal applications, Nambu et al. (2024) [32] reported the efficacy of tacrolimus combined with ustekinumab in steroid-refractory ulcerative colitis, and Guo et al. (2025) [33] developed RGD-HSA-TAC nanoparticles for diabetic kidney disease. Studies by Di Cocco et al. (2024) [34] and Braidotti et al. (2024) [35] assessed clinical outcomes of sublingual tacrolimus and early pediatric transplant exposure, respectively, offering alternative administration and age-specific strategies. From an immunological perspective, Kahn et al. (2024) [36] introduced a gene expression monitoring tool for tacrolimus-treated patients, while Nasa et al. (2024) [37] highlighted its combination with IL-2 in suppressing follicular helper T cells in lupus nephritis. Epigenetic insights were offered by Yusupov et al. (2024) [38] through DNA methylation studies of FKBP5. Additionally, Carretero-Ledesma et al. (2024) [39] and Krueger et al. (2024) [40] showed tacrolimus’s antiviral effects and influence on T-cell immunity against CMV. Chen et al. (2024) [41] explored hepatotoxicity mechanisms via JAK3/STAT3 signaling, and Ammar et al. (2024) [42] linked tacrolimus levels with oxidative stress markers. Studies by Gandon-Renard et al. (2024) [43] and Guo et al. (2024) [44] investigated FKBP12/12.6's role in cardiac and cancer models, indicating potential off-target effects and applications.
Tacrolimus pharmacokinetics has been extensively characterized, with key studies emphasizing its narrow therapeutic index, interindividual variability, and the critical need for personalized dosing strategies. Araya and Tasnif (2023) [45] highlighted its complex absorption, metabolism, and elimination pathways, laying the groundwork for further pharmacokinetic investigations. Genetic polymorphisms, particularly in CYP3A4, CYP3A5, and ABCB1, play a central role in variability, as shown by Alatorre-Moreno et al. (2024) [46], Vidal-Alabró et al. (2024) [47]. These studies collectively demonstrate that both donor and recipient genotypes significantly influence tacrolimus metabolism, clearance, and dose requirements. The impact of drug-drug interactions on tacrolimus levels has also been well documented. So et al. (2025) [48] identified interactions with cannabidiol, Boland et al. (2024) [49] with Nirmatrelvir/Ritonavir, Sun et al. (2024) [50] with deoxyschizandrin, Mahieu et al. (2024) [51] with rivaroxaban, and Miedziaszczyk et al. (2024) [52] with omeprazole/famotidine. Maruyama et al. (2024) [53] and Zhao et al. (2024) [54] further emphasized the importance of therapeutic drug monitoring when co-administered with letermovir and voriconazole, respectively. PBPK modelling has emerged as a powerful tool for predicting tacrolimus exposure in special populations: Xu et al. (2025) [22] in pregnant women, Guan et al. (2024) [23] in paediatric HSCT patients, and Martischang et al. (2024) [20] for optimizing IV perfusion and steady-state concentrations. Population pharmacokinetic models, such as those by Agergaard et al. (2025) [55] and Marco et al. (2024) [56], have been utilized to guide dose adjustments in kidney transplant recipients. Machine learning approaches by Li et al. (2024) [57] and real-world dosing optimization in Hispanic populations by Chamzas et al. (2024) [58] further support the trend toward individualized therapy. Xiang et al. (2024) [59] introduced salivary microbiota as a novel contributor to tacrolimus pharmacokinetics, while Tharanon et al. (2024) [60] and Ma et al. (2024) [61] reported biochemical and disease-related factors (e.g., bilirubin, haemoglobin, liver cancer) influencing clearance. Alternative formulations and delivery methods are also under exploration—Giral et al. (2024) [62] and Torres et al. (2024) [63] examined extended-release (LCPT) tacrolimus, noting benefits in tremor reduction and lower post-transplant diabetes risk. Momper et al. (2024) [64] evaluated MeltDose and in situ depot technologies for improving steady-state achievement and controlled release, respectively. Collectively, these studies underscore the intricate interplay of genetic, physiological, and external factors in tacrolimus pharmacokinetics and affirm the necessity of precision dosing to optimize therapeutic outcomes in transplant recipients.
Tacrolimus uptake from the digestive system after oral intake is incomplete and inconsistent. The total availability in grown kidney transplant individuals is 17±10%; in adult liver transplant cases is 22±6%; in well individuals is 18±5%. The overall absorption in young liver transplant cases was 31±24%. Tacrolimus highest blood measurements (Cmax) and region under the curve (AUC) seemed to grow in a dose-dependent pattern in 18 fasted well participants given a one-time oral dose of 3, 7, and 10 mg. When taken without nutrition, the speed and scope of uptake were the highest. The timing of eating also influenced availability. When consumed right after eating, mean Cmax decreased by 71%, and mean AUC dropped 39%, in comparison to the empty stomach condition. When provided 1.5 hours post-meal, mean Cmax fell 63%, and mean AUC lowered 39%, compared to the empty stomach state.
Tacrolimus exhibits varying volumes of distribution depending on patient condition and route of administration. In paediatric liver transplant patients, the volume of distribution is 2.6 ± 2.1 L/kg, while in individuals with renal impairment receiving 0.02 mg/kg every 4 hours intravenously (IV), it is 1.07 ± 0.20 L/kg. Patients with mild hepatic impairment show a distribution of 3.1 ± 1.6 L/kg for the same IV dose and 3.7 ± 4.7 L/kg when administered 7.7 mg orally (PO). In cases of severe hepatic impairment, the volume of distribution is 3.9 ± 1.0 L/kg for the IV dose and 3.1 ± 3.4 L/kg when given 8 mg orally. Tacrolimus is ~99% bound to human plasma proteins, primarily albumin and alpha-1-acid glycoprotein, with this binding remaining constant across concentrations ranging from 5 to 50 ng/mL.
Tacrolimus metabolism is primarily facilitated by CYP3A4 and to a lesser extent by CYP3A5, leading to the formation of eight metabolites, including 13-demethyl tacrolimus, 31-demethyl tacrolimus, 15-demethyl tacrolimus, 12-hydroxy tacrolimus, 15,31-didemethyl tacrolimus, 13,31-didemethyl tacrolimus, 13,15-didemethyl tacrolimus, and a final metabolite involving O-demethylation and fused ring formation. The predominant metabolite in human liver microsome incubations is 13-demethyl tacrolimus, while 31-demethyl tacrolimus retains similar activity to the parent compound.
In humans, less than 1% of the administered dose is excreted unchanged in urine. When given intravenously (IV), faecal elimination accounts for 92.6 ± 30.7%, whereas urinary elimination is 2.3 ± 1.1%. The elimination half-life varies across populations: 35 hours in healthy adults, 19 hours in kidney transplant patients, 12 hours in liver transplant patients, and 24 hours in heart transplant patients. In paediatric cases, the half-life is 11.5 ± 3.8 hours for liver transplant patients and 10.2 ± 5.0 hours (range 3.4–25 hours) for kidney transplant patients. The clearance rates also vary, with 0.040 L/hr/kg in healthy subjects (IV), 0.172 ± 0.088 L/hr/kg in healthy subjects (oral), 0.083 L/hr/kg in adult kidney transplant patients (IV), 0.053 L/hr/kg in adult liver transplant patients (IV), and 0.051 L/hr/kg in adult heart transplant patients (IV). In paediatric patients, clearance is 0.138 ± 0.071 L/hr/kg for liver transplant patients and 0.12 ± 0.04 L/hr/kg (range 0.06–0.17) for kidney transplant patients. Patients with renal impairment have a clearance of 0.038 ± 0.014 L/hr/kg (IV, 0.02 mg/kg/4 hr dose), while those with mild hepatic impairment show values of 0.042 ± 0.02 L/hr/kg (IV, 0.02 mg/kg/4 hr dose) and 0.034 ± 0.019 L/hr/kg (PO, 7.7 mg dose). In severe hepatic impairment, clearance further decreases to 0.017 ± 0.013 L/hr/kg (IV, 0.02 mg/kg/4 hr dose) and 0.016 ± 0.011 L/hr/kg (PO, 8 mg dose).
Adverse effects may be extreme and involve impaired eyesight, hepatic and renal complications (it is toxic to kidneys), convulsions, shaking, high blood pressure, low magnesium levels, sugar metabolism disorder, excess potassium, skin irritation, sleep disturbances, disorientation. Lethal dose (LD50) = 134-194 mg/kg (rodent).
Tacrolimus interacts with various analgesics, potentially altering its pharmacokinetics and increasing the risk of adverse effects. Acetaminophen can elevate tacrolimus serum concentrations, which may require dosage adjustments to prevent toxicity. Nonsteroidal anti-inflammatory drugs (NSAIDs) such as aceclofenac, acemetacin, acetylsalicylic acid, and diclofenac can heighten the risk of renal failure when co-administered with tacrolimus. Opioid analgesics, including alfentanil and fentanyl, may alter tacrolimus metabolism, potentially leading to increased drug levels. These interactions necessitate close monitoring to avoid complications such as nephrotoxicity, altered immunosuppression, and enhanced adverse effects.
Tacrolimus interacts with certain antianxiety drugs, influencing their metabolism and effects. When combined with buspirone, tacrolimus can decrease its metabolism, potentially increasing buspirone's sedative effects. Similarly, alprazolam experiences reduced metabolism when co-administered with tacrolimus, leading to prolonged sedation and an increased risk of side effects. Additionally, ketazolam and tacrolimus together result in elevated tacrolimus serum concentrations, which may require dose adjustments. These interactions highlight the need for careful monitoring when tacrolimus is used alongside antianxiety medications to avoid adverse effects and ensure therapeutic efficacy.
Tacrolimus interacts with antiarrhythmic medications, potentially leading to altered drug metabolism and increased risks of adverse effects. Amiodarone can elevate tacrolimus serum concentrations, increasing the likelihood of toxicity. Ajmaline, Amantadine, Amifampridine, Amiodarone, Amisulpride, Amodiaquine, Amoxapine, Antazoline, Arformoterol, Asenapine, Atomoxetine, Bepridil, Bretylium, Cinoxacin, Degarelix, Delamanid, Disopyramide, Dolasetron, Dronedarone, Droperidol, Entrectinib, Escitalopram, Flecainide, Ibutilide, Iloperidone, Lapatinib, Mefloquine, Mesoridazine, and Mexiletine can enhance the risk or severity of QTc prolongation, which may predispose patients to life-threatening arrhythmias. Digoxin excretion may be reduced when combined with tacrolimus, potentially leading to elevated plasma levels and toxicity. Given these interactions, careful monitoring of tacrolimus levels and dose adjustments are necessary when co-administered with antiarrhythmic agents to avoid serious complications.
Tacrolimus interacts with various antibacterial agents, affecting its metabolism, excretion, and overall pharmacological response. The serum concentration of tacrolimus can be increased when combined with chloramphenicol, ciprofloxacin, clarithromycin, erythromycin, and fluconazole, potentially leading to higher drug levels and an increased risk of toxicity. Amikacin, amphotericin B, and bacitracin may enhance the nephrotoxic effects of tacrolimus, increasing the likelihood of kidney damage. The excretion rate of amoxicillin, ampicillin, and cefdinir may be reduced when administered with tacrolimus, leading to higher systemic exposure of these antibiotics. Additionally, colistin and fosfomycin may also have decreased excretion when used in combination with tacrolimus. Close monitoring is recommended when co-administering tacrolimus with these antibacterials to prevent adverse effects and ensure therapeutic efficacy.
Tacrolimus interacts with various antibiotics, affecting its metabolism, serum concentration, and excretion. Macrolide antibiotics such as clarithromycin, erythromycin, and azithromycin can significantly increase tacrolimus serum levels by inhibiting its metabolism through the CYP3A4 enzyme, leading to a higher risk of toxicity. Fluoroquinolones like ciprofloxacin also increase tacrolimus levels, requiring careful dose adjustments. On the other hand, rifampin (a rifamycin antibiotic) induces CYP3A4, reducing tacrolimus levels, which may necessitate dose escalation. Beta-lactam antibiotics, including amoxicillin and ampicillin, may decrease tacrolimus excretion, leading to elevated serum levels. Aminoglycosides such as amikacin and gentamicin can impair nephrotoxicity when combined with tacrolimus. Additionally, antifungal agents like fluconazole and voriconazole can also increase tacrolimus concentration, requiring close monitoring. These interactions highlight the importance of therapeutic drug monitoring when prescribing antibiotics alongside tacrolimus to prevent toxicity or subtherapeutic effects.
Tacrolimus interacts with several anticoagulants and thrombolytics, increasing the risk or severity of bleeding. When combined with Abciximab, Alteplase, Ancrod, Anistreplase, Antithrombin Alfa, Argatroban, Bivalirudin, Dabigatran, Dabigatran etexilate, Defibrotide, Desirudin, Dipyridamole, Drotrecogin alfa, Edetic acid, Edoxaban, Epoprostenol, Eptifibatide, and Fluindione, tacrolimus can enhance their anticoagulant or thrombolytic effects, leading to an elevated risk of haemorrhagic complications. These interactions necessitate close monitoring of coagulation parameters and potential dose adjustments to mitigate bleeding risks.
Tacrolimus interacts with several anticonvulsants, influencing its metabolism and serum concentration. The metabolism of tacrolimus can be increased when combined with topiramate, potentially leading to reduced drug efficacy. Similarly, primidone and probenecid enhance tacrolimus metabolism, which may necessitate dosage adjustments. On the other hand, lacosamide and lamotrigine can increase the risk or severity of hyperkalemia when administered with tacrolimus, posing potential electrolyte imbalances. Additionally, brivaracetam may decrease the excretion rate of tacrolimus, leading to elevated drug levels and an increased risk of toxicity.
Selective Serotonin Reuptake Inhibitors (SSRIs) such as fluoxetine, fluvoxamine, and sertraline may increase tacrolimus levels by inhibiting CYP3A4, potentially leading to toxicity, nephrotoxicity, or neurological side effects. Tricyclic antidepressants (TCAs) like amitriptyline may also affect tacrolimus metabolism, requiring dose adjustments. Monoamine oxidase inhibitors (MAOIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) such as venlafaxine and duloxetine could alter tacrolimus levels, increasing the risk of adverse effects like hypertension or neurotoxicity. Additionally, tacrolimus itself may enhance the neurotoxic effects of some antidepressants, leading to symptoms like tremors, confusion, or seizures.
Tacrolimus interacts with various antifungal agents, affecting its metabolism and serum concentration. Ketoconazole, clotrimazole, and fluconazole significantly increase tacrolimus levels by inhibiting CYP3A4, leading to a higher risk of toxicity. Itraconazole and voriconazole also elevate tacrolimus concentrations, requiring dose adjustments to avoid nephrotoxicity and other adverse effects. Amphotericin B, when co-administered with tacrolimus, enhances nephrotoxicity, necessitating careful renal function monitoring. On the other hand, caspofungin decreases tacrolimus levels, potentially reducing its immunosuppressive efficacy. Proper dose modifications and therapeutic drug monitoring are essential when combining tacrolimus with antifungal agents to prevent toxic effects or therapeutic failure.
Tacrolimus interacts with antihistamines by influencing their metabolism and potential side effects. When combined with antihistamines such as cetirizine, hydroxyzine, acrivastine, astemizole, chlorpheniramine, desloratadine, dexchlorpheniramine maleate, and triprolidine, the risk or severity of QTc prolongation increases. This means that the combination can potentially lead to dangerous cardiac arrhythmias. Additionally, tacrolimus may decrease the metabolism of azelastine and chlorpheniramine, leading to increased serum levels of these drugs and potentially amplifying their sedative and systemic effects.
Tacrolimus interacts with several antihypertensive medications, potentially altering their effects or leading to adverse reactions. For example, combining tacrolimus with angiotensin-converting enzyme (ACE) inhibitors like benazepril, enalapril, and fosinopril can increase the risk or severity of hyperkalemia, a condition characterized by elevated potassium levels, which can be dangerous for patients with compromised kidney function. Similarly, angiotensin II receptor blockers (ARBs) such as candesartan and eprosartan may also enhance the risk of hyperkalemia when taken with tacrolimus. Beta-blockers like atenolol, bisoprolol, and celiprolol may have their serum concentration increased, potentially leading to excessive cardiovascular effects, while calcium channel blockers such as amlodipine and felodipine can enhance the immunosuppressive effects of tacrolimus. Additionally, loop and thiazide diuretics, including bumetanide and bendroflumethiazide, may increase the risk of neutropenia and thrombocytopenia, conditions that can weaken the immune system. These interactions highlight the importance of careful monitoring and dose adjustments when tacrolimus is administered alongside antihypertensive medications.
Tacrolimus interacts significantly with anti-inflammatory drugs, particularly nonsteroidal anti-inflammatory drugs (NSAIDs) such as Aceclofenac, Acemetacin, Acetylsalicylic acid, Alclofenac, Aminophenazone, Antrafenine, Benorilate, Benoxaprofen, Balsalazide, Carprofen, Celecoxib, Dexibuprofen, Dexketoprofen, Diclofenac, Diflunisal, Etodolac, Fenbufen, Fenoprofen, Flurbiprofen, and Indomethacin. When combined with these medications, there is an increased risk of renal failure, as both tacrolimus and NSAIDs can impair kidney function by reducing blood flow to the kidneys. Additionally, drugs like acetaminophen may elevate tacrolimus serum concentrations, potentially increasing the risk of toxicity. Due to these interactions, close monitoring of kidney function and tacrolimus levels is essential when co-administering these medications.
Tacrolimus interacts with several antineoplastic agents, primarily by enhancing their immunosuppressive effects. When combined with Cisplatin, Cladribine, Belinostat, Carboplatin, Bendamustine, Capecitabine, Carmustine, Carfilzomib, Gemcitabine, and Gemtuzumab ozogamicin, tacrolimus may increase their immunosuppressive activity, potentially leading to heightened risks of infections and immune suppression. Additionally, interactions with Caplacizumab and Cangrelor may elevate the risk of bleeding, necessitating close monitoring of coagulation parameters. These interactions highlight the need for careful dosage adjustments and monitoring when tacrolimus is used alongside antineoplastics to mitigate adverse effects while maintaining therapeutic efficacy.
Tacrolimus interacts with antipsychotic medications in various ways, affecting both drug metabolism and potential side effects. The metabolism of certain antipsychotics, such as aripiprazole and its prodrug aripiprazole lauroxil, can be decreased when combined with tacrolimus, potentially leading to increased plasma levels and prolonged drug effects. Similarly, quetiapine metabolism may be reduced, increasing the risk of sedation and other side effects. Clozapine, another antipsychotic, can have increased serum concentrations when administered with tacrolimus, potentially leading to an enhanced risk of adverse effects. Additionally, some antipsychotics, such as olanzapine and asenapine, may increase the risk of QTc prolongation when combined with tacrolimus, raising concerns about potential cardiac arrhythmias. The combination of tacrolimus with haloperidol has also been shown to increase tacrolimus serum concentrations, possibly necessitating dose adjustments. These interactions highlight the need for careful monitoring of therapeutic levels and side effects when tacrolimus is used alongside antipsychotic medications.
Tacrolimus interacts with antipyretics in ways that can impact drug metabolism and patient safety. Acetaminophen, a commonly used antipyretic, can increase the serum concentration of tacrolimus, potentially leading to enhanced immunosuppressive effects and a higher risk of toxicity. Additionally, acetylsalicylic acid (aspirin), another widely used antipyretic, may increase the risk or severity of renal failure when combined with tacrolimus. These interactions suggest that careful monitoring of tacrolimus levels is necessary when co-administering antipyretics, especially in patients with compromised kidney function or those on high doses of tacrolimus.
Tacrolimus interacts with various antivirals, leading to changes in drug metabolism and potential adverse effects. When combined with Acyclovir, the risk or severity of nephrotoxicity increases, necessitating close renal function monitoring. Similarly, Adefovir dipivoxil also elevates the likelihood of nephrotoxic effects, which could compromise kidney function. Amprenavir and Atazanavir significantly increase the serum concentration of tacrolimus, potentially leading to toxicity if not properly adjusted. Boceprevir and Darunavir also elevate tacrolimus levels, requiring dosage modifications. The metabolism of Tacrolimus can be decreased when combined with Daclatasvir, leading to prolonged drug activity. Foscarnet, like Acyclovir and Adefovir, raises the risk of nephrotoxicity, necessitating careful dose regulation and hydration strategies. Delavirdine, Efavirenz, and Elvitegravir may also increase tacrolimus exposure, necessitating therapeutic monitoring to avoid toxicity. Ritonavir-boosted regimens, such as those including Lopinavir or Saquinavir, significantly impact tacrolimus metabolism, leading to dose adjustments to maintain therapeutic levels. Given these interactions, patients on tacrolimus therapy who require antiviral treatment should undergo regular therapeutic drug monitoring to optimize efficacy and minimize adverse effects.
Tacrolimus interacts with beta-blockers by potentially increasing the risk or severity of hyperkalemia. Beta-blockers such as atenolol, acebutolol, penbutolol, metoprolol, betaxolol, and labetalol can contribute to elevated potassium levels when used concomitantly with tacrolimus. This interaction may lead to complications, especially in patients with renal impairment or those on additional potassium-retaining medications. Monitoring potassium levels and renal function is advised when these drugs are prescribed together to mitigate the risk of hyperkalemia and associated adverse effects.
Tacrolimus has potential interactions with bronchodilators, particularly albuterol (salbutamol) and arformoterol, which may increase the risk of QTc prolongation when combined. This can lead to arrhythmias or other cardiovascular complications, making careful monitoring necessary in patients requiring both medications. Given tacrolimus's narrow therapeutic index, healthcare providers should be vigilant about cardiac risks, especially in individuals with pre-existing heart conditions. Adjustments in dosing or alternative treatment strategies may be considered to mitigate these potential effects.
Tacrolimus interacts with various cytotoxic agents, potentially influencing their efficacy and toxicity. It may enhance the immunosuppressive effects of several cytotoxic drugs, including carboplatin, carfilzomib, and carmustine. This increased immunosuppression can elevate the risk of opportunistic infections and malignancies in patients undergoing chemotherapy. Additionally, tacrolimus co-administration with cytarabine, dacarbazine, dactinomycin, and danicopan may further potentiate immunosuppressive activities, necessitating careful monitoring. Given the risks associated with these interactions, clinicians should adjust dosing regimens and monitor patients for potential adverse effects.
Tacrolimus interacts with various hormones, affecting their metabolism and efficacy. When combined with epinephrine, the serum concentration of tacrolimus can increase, potentially amplifying its immunosuppressive effects. Estradiol and its derivatives, including estradiol acetate, benzoate, cypionate, dienanthate, and valerate, can alter the metabolism of tacrolimus, by either increasing or decreasing its breakdown, which may affect therapeutic levels. Testosterone undecanoate can also elevate tacrolimus concentrations, which may necessitate dose adjustments to prevent toxicity. Additionally, hydrocortisone and its various forms, such as acetate, butyrate, cypionate, phosphate, probutate, succinate, and valerate, may enhance tacrolimus’s immunosuppressive activity, increasing the risk of infections. Other hormones like somatotropin, somatrogon, and histrelin can influence tacrolimus pharmacodynamics, with somatotropin potentially leading to myopathy, rhabdomyolysis, and myoglobinuria, and histrelin increasing the risk of QTc prolongation. These interactions highlight the need for careful monitoring when tacrolimus is administered alongside hormonal therapies.
Tacrolimus can interact with oral hypoglycemic agents, potentially reducing their therapeutic efficacy. For instance, drugs like Acarbose, Acetohexamide, Alogliptin, Albiglutide, Canagliflozin, Chlorpropamide, Dapagliflozin, Dulaglutide, Empagliflozin, Ertugliflozin, and Exenatide may have diminished glucose-lowering effects when co-administered with tacrolimus. This interaction could lead to poorer blood sugar control in diabetic patients, necessitating careful monitoring and potential dose adjustments of the hypoglycemic agents. Healthcare providers should assess blood glucose levels regularly in patients receiving both tacrolimus and oral hypoglycemics to ensure optimal glycemic management.
Tacrolimus interacts with various immunosuppressive agents, altering their efficacy and potential side effects. It may enhance the immunosuppressive activity of drugs like Abatacept, Adalimumab, Alefacept, Alemtuzumab, Anakinra, Anifrolumab, Antithymocyte immunoglobulin (rabbit), Apremilast, Azathioprine, Basiliximab, Beclomethasone dipropionate, Belatacept, Belimumab, Bimekizumab, Brodalumab, Budesonide, Canakinumab, Certolizumab pegol, Clobetasol, Clobetasone, Corticotropin, Deflazacort, Dimethyl fumarate, Etanercept, Fingolimod, Fluticasone, Mycophenolate mofetil, Prednisolone, Rituximab, and Sirolimus. These interactions can lead to an increased risk of infections and complications related to excessive immunosuppression. Additionally, Tacrolimus in combination with Cyclosporine may heighten nephrotoxicity, necessitating close monitoring of renal function. It is crucial to adjust dosages and monitor patients carefully when tacrolimus is used alongside other immunosuppressive medications to optimize therapeutic outcomes while minimizing adverse effects.
Tacrolimus interacts with various narcotics, influencing their metabolism, serum concentration, and excretion. When combined with Buprenorphine, the serum concentration of tacrolimus increases, potentially leading to higher drug exposure. Oxycodone metabolism is reduced in the presence of tacrolimus, which may prolong its effects and increase the risk of toxicity. Similarly, Hydromorphone excretion is slowed down when taken with tacrolimus, leading to prolonged drug action. Opium also experiences decreased excretion, which could amplify its pharmacological effects. Additionally, Morphine concentration in the serum can be elevated when administered alongside tacrolimus, potentially intensifying its analgesic and side effects.
Tacrolimus interacts with various sedatives, affecting their metabolism and excretion. When combined with diazepam, the serum concentration of tacrolimus increases, potentially leading to enhanced sedative effects. Similarly, tacrolimus may decrease the excretion rate of dexmedetomidine, which could result in prolonged sedation. The metabolism of buspirone is also decreased when used alongside tacrolimus, leading to higher drug levels and an increased risk of central nervous system depression. Additionally, tacrolimus can reduce the excretion rate of flurazepam, potentially leading to prolonged sedation and drowsiness. These interactions highlight the need for careful monitoring and dose adjustments when using tacrolimus with sedatives to prevent excessive sedation or adverse effects.
Tacrolimus interacts with various female sex hormones, affecting their metabolism and excretion. The metabolism of tacrolimus can be decreased when combined with estradiol, potentially leading to increased serum concentrations. Similar effects have been observed with estradiol acetate, estradiol benzoate, estradiol cypionate, estradiol dienanthate, and estradiol valerate, where their metabolism is enhanced in the presence of tacrolimus, altering hormonal levels in the body. Additionally, estrone sulfate may experience a reduced excretion rate when administered alongside tacrolimus, resulting in prolonged systemic exposure. Furthermore, synthetic conjugated estrogens (A and B) exhibit a reduced excretion rate when co-administered with tacrolimus, which could impact hormonal balance and therapeutic efficacy.
The patients receiving tacrolimus must not drink alcohol, it can boost the speed of tacrolimus delivery. Grape fruit and other citrus fruits should also be avoided due their potential effect on its metabolism. St. John's Wort also enhances the CYP3A4 breakdown of tacrolimus; thus, checking tacrolimus whole blood trough figures could be necessary. Further tacrolimus should be taken at identical time daily without meals, at minimum 1 hour ahead or 2 hours following food. Taking together with meals, reduces the rate and extent of absorption and on the other hand, administration with stomach acid neutralizers such as aluminium or magnesium hydroxide antacids could elevate tacrolimus levels, raising concerns about overdose.
Pharmacogenomics explores how genetic variations influence drug response, playing a crucial role in optimizing tacrolimus therapy. Tacrolimus pharmacokinetics and response are influenced by genetic polymorphisms, primarily involving CYP3A4, CYP3A5, ABCB1 (MDR1), POR (P450 Oxidoreductase), and NR1I2 (PXR). These genes regulate tacrolimus metabolism, transport, and intracellular distribution, significantly impacting drug dosing and patient outcomes. Understanding these genetic factors enables personalized dosing strategies, improving therapeutic outcomes and minimizing adverse effects in transplant patients.
Concha et al. (2024) [66] discovered that the CYP3A4*1B polymorphism played a significant role in long-term tacrolimus dose requirements in Spanish solid organ transplant patients. Carriers of CYP3A4*1B required higher doses and exhibited lower blood concentrations, independent of the influence of CYP3A5*3.
Nakamura et al. (2020) [67] reported that the **CYP3A53/3 genotype significantly influenced the tacrolimus concentration/dose ratio in liver transplant patients, with POR*28 also contributing to variability. Similarly, Contreras-Castillo et al. (2021) [68] found that **CYP3A53/3 carriers required lower tacrolimus doses and exhibited higher dose-adjusted trough levels in Chilean kidney transplant patients. Srinivas et al. (2021) [69] highlighted that CYP3A5 expressors had a greater risk of transplant rejection, whereas non-expressors were more prone to developing new-onset diabetes mellitus post-transplantation. Cheng et al. (2021) [70] identified CYP3A5*3 as the primary determinant of tacrolimus dose-adjusted trough concentrations in kidney transplant recipients, whereas CYP3A4 and ABCB1 polymorphisms were not significantly associated. Finally, Marco et al. (2024) [71] demonstrated that the CYP3A5 genotype influenced tacrolimus pharmacokinetics and was linked to the incidence of graft-versus-host disease in hematopoietic stem cell transplantation.
Tron et al. (2020) [72] reported that the ABCB1 1199G>A polymorphism was associated with variability in tacrolimus whole blood and intracellular exposure, while CYP3A4 and CYP3A5 did not show significant correlations. Similarly, Bonezi et al. (2020) [73] found that the ABCB1 C3435T and G2677T polymorphisms influenced tacrolimus dose requirements, with T alleles being linked to an increased risk of acute rejection in transplant patients.
Nakamura et al. (2020) [67] found that the POR*28 polymorphism significantly reduced the tacrolimus blood concentration/dose ratio in liver transplant recipients carrying the CYP3A5*1 allele. Similarly, Cheng et al. (2021) [74] reported that POR*28 was associated with lower dose-adjusted tacrolimus concentrations, particularly when used in combination with cyclosporine.
Yamamoto et al. (2020) [75] found that NR1I2 (PXR) genetic variants influenced tacrolimus intracellular distribution in liver transplant patients, impacting their pharmacodynamic responses.
CONCLUSION
Tacrolimus being a narrow therapeutic index drug with high pharmacokinetic variability and associated drug interactions, it is still widely used immunosuppressant for prevention of organ transplant rejections. These factors necessitate, its regular therapeutic drug monitoring together with dose adjustment, hence posing great difficulties for clinicians and researchers involved in the studies on this drug. It, therefore becomes important for clinicians and researchers to know the factors that influence its pharmacokinetics and pharmacodynamics and thereby reducing the associated unfavourable outcomes. The information on pharmacokinetics, pharmacodynamics, genetic polymorphism and drug-drug interactions of tacrolimus would assist the physicians to optimize and individualize the drug therapy.
REFERENCES

Sajad Khaliq Dar, Arshad H. Khuroo, Mohd. Akhtar*, Sudershan Kumar, Chandni Sharma, Tacrolimus: An Updated Review on Pharmacological, Pharmacodynamic, Pharmacokinetic Profile and Pharmacogenomics, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 4, 2873-2893. https://doi.org/10.5281/zenodo.15271719
 10.5281/zenodo.15271719
10.5281/zenodo.15271719
