Drug design is a multidisciplinary endeavor focused on the discovery and development of novel therapeutic agents with optimized efficacy, safety, and pharmacokinetic properties. It synthesizes principles from medicinal chemistry, pharmacology, molecular biology, and computational modeling to identify and refine lead compounds. Modern approaches include structure-based drug design (SBDD), which leverages the three-dimensional structures of biological targets, and ligand-based drug design (LBDD), which utilizes the chemical features of known active compounds. Recent advances in computational methods, high-throughput screening, and artificial intelligence have significantly accelerated the processes of lead identification and optimization. Iterative refinement is employed to improve properties such as binding affinity, selectivity, solubility, and metabolic stability, while minimizing off-target effects and toxicity. Ultimately, drug design plays a critical role in bridging the gap between target identification and clinical application, driving the development of innovative therapeutics for a wide range of diseases.
Drug Design, SBDD, LBDD, CADD, PubChem, Docking, ADMET
Drug design is a systematic and strategic process aimed at discovering and developing new pharmaceutical agents that can precisely interact with specific biological targets to treat or prevent diseases. It acts as a vital bridge between fundamental biomedical research and clinical application, with the goal of producing molecules that exhibit high specificity and potency, along with favorable pharmacokinetic properties and safety profiles.
The field primarily involves two main approaches: structure-based drug design (SBDD), which utilizes the three-dimensional structure of the target biomolecule to guide compound design, and ligand-based drug design (LBDD), which relies on knowledge of existing biologically active molecules to create new candidates. Advances in computational chemistry, molecular modelling, and bioinformatics have greatly improved the efficiency and accuracy of both methods.[1] Modern drug design integrates in silico techniques such as molecular docking, quantitative structure–activity relationship (QSAR) modelling, and pharmacophore mapping to predict interactions between molecules and their targets. These methods, combined with high-throughput screening and combinatorial chemistry, have accelerated the discovery process, reduced costs, and increased the likelihood of identifying successful drug candidates. Overall, drug design represents the convergence of chemistry, biology, and computational science, driving innovation in the creation of targeted therapeutics for a wide range of health conditions.[2]. Drug design is a multidisciplinary process aimed at identifying and developing new therapeutic agents that can specifically interact with biological targets to treat, manage, or prevent diseases. It represents a crucial stage in the drug discovery pipeline, transforming basic biological and chemical knowledge into practical medicinal solutions. The process seeks to create molecules that possess optimal binding affinity, high selectivity, minimal toxicity, and favourable pharmacokinetic properties such as absorption, distribution, metabolism, and excretion (ADME).[3]. In recent decades, technological advancements have significantly transformed the field. Computational tools now play a central role, enabling in silico screening of vast chemical libraries, prediction of molecular interactions, and optimization of lead compounds through molecular docking, pharmacophore modelling, and quantitative structure–activity relationship (QSAR) analysis. These approaches, combined with high-throughput screening, combinatorial chemistry, and artificial intelligence, have accelerated the pace of discovery, reduced research costs, and increased the probability of clinical success.[4]. Drug design also involves iterative refinement, where candidate molecules are continuously modified to enhance desired properties and eliminate drawbacks. This process requires close integration between medicinal chemistry, pharmacology, toxicology, and formulation science. Ultimately, successful drug design not only contributes to medical innovation but also addresses unmet healthcare needs, paving the way for the development of next-generation therapeutics targeting complex and challenging diseases.[5]
History and Evolution of Drug Design
The history of drug design can be traced back to ancient civilizations, where medicinal preparations were derived directly from natural sources such as plants, minerals, and animal products. Early drug discovery was based largely on trial-and-error methods, traditional knowledge, and empirical observations rather than a scientific understanding of disease mechanisms. Herbal remedies, alkaloids, and crude extracts formed the basis of ancient pharmacotherapy. The 19th century marked the beginning of the scientific era of drug discovery. The isolation of pure compounds, such as morphine from opium and quinine from cinchona bark, allowed for the precise study of chemical structures and pharmacological effects. The development of organic chemistry during this period enabled scientists to synthesize new molecules and modify existing natural compounds for improved therapeutic properties.[6] In the early 20th century, the concept of “magic bullets” proposed by Paul Ehrlich revolutionized drug development. Ehrlich envisioned designing chemicals that could selectively target disease-causing agents without harming healthy tissues, laying the foundation for targeted drug design. This era saw the discovery of sulphonamides and the introduction of antibiotics like penicillin, which transformed medicine by providing effective treatments for infectious diseases. The mid-20th century brought the rise of rational drug design, supported by advancements in biochemistry and molecular biology. Scientists began to understand enzymes, receptors, and other biomolecules as drug targets. The discovery of DNA’s structure in 1953 and the growth of molecular pharmacology paved the way for drugs designed to interact with specific biological pathways.[7]. From the late 20th century onwards, the integration of computational methods revolutionized drug design. Structure-based drug design (SBDD) emerged as a powerful tool, using three-dimensional structural information obtained from X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy. Ligand-based drug design (LBDD) also advanced, relying on data from known active compounds to design analogs with improved properties. In the 21st century, drug design has entered an era of precision and personalization. Computational chemistry, molecular docking, high-throughput screening, and artificial intelligence are now integral parts of the process. These technologies allow rapid screening of millions of compounds, prediction of pharmacokinetic and toxicity profiles, and optimization of drug candidates before synthesis. Additionally, advances in genomics and proteomics have paved the way for personalized medicine, where therapies can be tailored to an individual’s genetic makeup and disease profile.[8]
Introduction to CADD
Computer-Aided Drug Design (CADD) is a modern approach in pharmaceutical research that utilizes computational tools and modelling techniques to identify, design, and optimize potential drug candidates. It plays a crucial role in reducing the time, cost, and resources required for traditional drug discovery by allowing scientists to simulate and analyze molecular interactions before laboratory synthesis and testing.[9] CADD integrates principles from medicinal chemistry, molecular biology, bioinformatics, and structural biology to predict how a compound will interact with a specific biological target. By employing mathematical models, molecular docking, virtual screening, and quantitative structure–activity relationship (QSAR) analyses, researchers can assess the binding affinity, selectivity, and pharmacokinetic properties of compounds in silico.
There are two main categories of CADD:
- Structure-Based Drug Design (SBDD) – Relies on the 3D structure of the target protein to design molecules that fit precisely into its binding site.
- Ligand-Based Drug Design (LBDD) – Used when the target structure is unknown but data on active molecules is available; this method designs new compounds by analyzing the chemical and biological properties of known ligands.[10]
Chemical Structure Representation
Chemical structure representation is the method of illustrating the arrangement of atoms in a molecule and the chemical bonds between them. It serves as a visual and logical way to understand a compound’s composition, geometry, and reactivity. Representations can be two-dimensional (2D) for structural clarity or three-dimensional (3D) for spatial and conformational analysis.
1. Types of Chemical Structure Representation
a) Molecular Formula
- Shows the total number and types of atoms in the molecule.
- Example: C?H??N?O? (Caffeine).
- Limitation: Does not indicate atom connectivity or geometry.
b) Lewis Structure
- Displays atoms, valence electrons, and bonding patterns using dots and lines.
- Useful for understanding electron distribution and predicting reactivity.
c) Structural Formula (2D Representation)
- Full Structural Formula – Shows all atoms and bonds explicitly.
- Condensed Formula – Groups similar atoms together (e.g., CH?CH?OH for ethanol).
d) Skeletal (Line-Angle) Formula
- Widely used in organic chemistry.
- Carbon atoms are represented by line ends or vertices, and hydrogen atoms attached to carbon are omitted for simplicity.
- Example: Benzene is drawn as a hexagon with alternating double bonds.[2]
e) Ball-and-Stick Model (3D Representation)
- Atoms are spheres, bonds are sticks.
- Shows spatial arrangement and bond angles.
- Useful for molecular geometry visualization.
f) Space-Filling Model
- Atoms are represented by spheres scaled to their van der Waals radii.
- Gives an idea of molecular volume and surface properties.
g) Computer-Aided Models (CADD)
- 3D models generated using computational tools for drug design.
- Allows molecular docking, conformational analysis, and property prediction.
2. Importance in Drug Design and CADD
- Helps in understanding molecular interactions with biological targets.
- Essential for predicting binding affinity and drug efficacy.
- Facilitates virtual screening and structure optimization.
Chemical Database Search
A chemical database search is the process of retrieving information about chemical compounds, their properties, and related data from specialized digital repositories. These databases are essential tools in drug discovery, chemical research, and CADD (Computer-Aided Drug Design), enabling scientists to identify known compounds, predict properties, and explore potential new molecules.
1. Types of Chemical Databases
a) Structure Databases
Contain detailed molecular structures with bonding information.
Examples:
- PubChem – Public database by NCBI with millions of compounds.
- ChemSpider – Aggregates chemical data from multiple sources.
b) Spectral Databases
Store experimental spectra such as IR, NMR, MS.
Examples:
- NIST Chemistry WebBook – IR, MS, UV spectra.
- SDBS (Spectral Database for Organic Compounds) – NMR and MS data.
c) Bioactivity Databases
Provide data on biological activity of compounds against various targets.
Examples:
- ChEMBL – Bioactive drug-like molecules.
- BindingDB – Binding affinity data for protein–ligand interactions.
d) Commercial Databases
Require paid access; often have advanced search tools and proprietary data.
Examples:
- SciFinder – Comprehensive literature and chemical property database.
- Reaxys – Reaction and substance information from patents and journals.[5]
2. Search Methods in Chemical Databases
- Text Search – Using compound names, synonyms, CAS numbers.
- Structure Search – Drawing or uploading molecular structures for substructure, similarity, or exact match searches.
- Formula Search – Based on elemental composition.
- Property Search – Using parameters such as molecular weight, melting point, or logP.
- Spectral Search – Matching uploaded spectra against database records.
3. Applications in Drug Design
- Identification of known drugs or lead compounds for modification.
- Retrieval of ADME/Toxicity data for computational modeling.
- Exploration of patent literature to ensure novelty.
- Support for virtual screening and QSAR studies in CADD.
Nice — I’ll unpack each pharmacophore element and then explain how models are built, represented, validated, and used in practice. I’ll keep it practical (what it means physically, how it’s encoded in software, and what to watch out for).[3]
Key pharmacophore features — detailed
Hydrogen-bond donors (HBD)
- What they are: Functional groups that can donate a hydrogen to form an H-bond (common examples: –OH, –NH, –NH?).
- Why they matter: H-bonds contribute directionally to binding specificity and often determine the precise orientation of a ligand in a pocket.
- How they’re represented: As a feature point (often with a vector indicating H→acceptor directionality) plus a tolerance radius. Models may require both a distance and an angle (near linearity preferred) for good H-bond geometry.
- Modelling notes: Protonation and tautomer states matter — an –NH may be a donor or not depending on pH/tautomer; generate relevant protonation states and conformers.
Hydrogen-bond acceptors (HBA)
- What they are: Atoms or groups that accept H-bonds (carbonyl O, ether O, tertiary amine lone pairs).
- Why they matter: Complementary to donors; they often anchor ligands via polar interactions and can govern selectivity.
- How they’re represented: As points (sometimes with a direction vector if geometry matters), with radii for acceptable placement.
- Modelling notes: Some atoms can be both HBA and HBD depending on context (e.g., amides) — represent accordingly.
Aromatic/π systems
- What they are: Centers of delocalized π electrons (phenyl, heteroaromatics).
- Why they matter: Enable π–π stacking, T-shaped interactions, edge-to-face interactions, and cation–π contacts — important for affinity and orientation.
- How they’re represented: Usually as an aromatic centroid/plane feature (center + normal vector) and a radius (centroid-to-centroid tolerances). Some models capture ring plane orientation for stacking vs. T-shaped geometry.
- Modelling notes: Heteroaromatic rings may also contribute H-bonding or polarity, so features can overlap.
Positive / Negative ionizable groups (charged centres)
- 1. What they are: Protonated amines (cationic) or deprotonated acids (anionic) that form salt bridges, ionic interactions, or strong electrostatic contacts.
- 2. Why they matter: Ionic interactions can be among the strongest specific interactions in binding. Correct protonation state is critical.
- 3. How they’re represented: As charged feature points (cationic/anionic) with no directional requirement but with distance tolerances. Some tools also model salt-bridge geometry or electrostatic potential maps.
- 4. Modeling notes: Always check pKa and generate the likely microspecies at assay pH (often ~7.4). Wrong protonation → false negatives/positives.
Metal-binding features
- What they are: Groups that coordinate metal ions in the binding site (carboxylates, thiols, imidazoles coordinating Zn²?, Mg²?, etc.).
- Why they matter: Metal coordination often defines binding mode and requires specific geometry (tetrahedral, octahedral).
- How they’re represented: As special metal-binding features with geometry constraints and sometimes as explicit metal centers in the pharmacophore.
- Modelling notes: Include the metal explicitly in structure-based models; treat geometry constraints carefully.
Halogen-bonding / special interactions (optional advanced features)
- What they are: Directional interactions where a halogen (Cl, Br, I) uses its σ-hole to interact with an electron donor.
- Why they matter: Can be important for affinity and orientation in halogenated ligands.
- How they’re represented: As directional halogen bond features (vector + radius).
- Modelling notes: Not all tools model these; include only when halogen chemistry is present.
Excluded volumes (steric constraints)
- What they are: Spheres or shapes that mark regions in space where ligand atoms must not occupy (protein atoms/side chains).
- Why they matter: Prevent unrealistic placements and improve selectivity of virtual screening by enforcing steric complementarity.
- How they’re represented: As negative-space volumes derived from protein structure (important in structure-based pharmacophores).
- Modelling notes: Useful to reduce false positives; create from a reliable protein structure and consider mobility of side chains.
Types of pharmacophore models — how they differ
Ligand-based pharmacophores
- Built from: A set of known active ligands (aligned in 3-D).
- Method: Generate conformers of actives → overlay and extract common features → form hypothesis of shared features.
- Strengths: Useful when no protein structure is available; captures SAR implicit in actives.
- Limitations: Quality depends on the diversity and correctness of the input actives and on conformer sampling. Risk of overfitting to a small set.
Structure-based pharmacophores
- Built from: Protein 3-D structure (apo or with bound ligand).
- Method: Map interaction hotspots in the binding pocket (hydrogen-bonding sites, hydrophobic pockets, metal sites, waters) and translate into features + excluded volumes.
- Strengths: More mechanistic and can include steric constraints and water/metal interactions.
- Limitations: Dependent on protein structure quality and on capturing protein flexibility.
Ensemble / hybrid pharmacophores
- Combine multiple protein conformations or multiple ligand sets to model binding site flexibility and different binding modes. Useful to avoid missing actives due to a single static snapshot.
Typical workflow to create and use a pharmacophore model
- Collect high-quality data — active ligands, protein crystal structures (if available).
- Prepare ligands & protein — correct protonation, tautomers, 3-D geometry, remove artifacts.
- Generate conformers for each ligand to sample accessible geometries.
- Map features (automatically or manually) — identify HBD/HBA, hydrophobic, aromatic, charged spots.[4]
- Generate hypotheses — create one or several pharmacophore models (vary mandatory vs optional features).
- Validate models — test with known actives and decoys (enrichment metrics, ROC curves, early enrichment).
- Virtual screen chemical libraries — map each compound’s conformers to the pharmacophore and score matches.
- Post-filter & prioritize — docking, rescoring, ADME/Tox filters, or medicinal chemistry triage.
- Iterate — refine model with new actives or structural data.
Validation & performance metrics (practical)
- Enrichment factor (EF) — how many actives are found among top ranked hits versus random expectation (often reported at top 1%, 5%).
- ROC-AUC — overall ability to separate actives from inactives.
- Early enrichment metrics (e.g., BEDROC) — emphasize retrieval of actives at the very top of the ranked list (important for screening).
- Decoy sets: Use carefully chosen decoys (physiochemically similar but inactive) to avoid inflated performance.
- External test set: Always test on compounds not used to build the model.
Practical tips & common pitfalls
- Don’t over-constrain your pharmacophore. Too many mandatory features = almost zero hits. Use optional features where possible.
- Account for protonation & tautomerism. Wrong states → wrong mapping. Generate plausible states at assay pH.
- Conformer sampling is crucial. Missing a bioactive conformer causes false negatives.
- Include excluded volumes in structure-based models to reduce steric false positives.
- Beware of overfitting. Build models from diverse actives and validate on external sets.
- Combine methods. Follow pharmacophore screening with docking and ADMET filters for a robust hit list.
- Consider water molecules & metal ions explicitly when they mediate important interactions.
- Use ensemble approaches to capture protein flexibility if the binding pocket is plastic.
Common tools (short list)
- LigandScout, Phase (Schrödinger), MOE, Discovery Studio, PharmaGist, Pharmer — most support ligand- and/or structure-based pharmacophore modeling and virtual screening.
Docking
Docking is a computational technique used in Computer-Aided Drug Design (CADD) to predict how a small molecule (ligand) binds to a specific site on a macromolecular target (such as a protein or enzyme). Its goal is to determine the optimal binding orientation and estimate the strength of the interaction between the ligand and the target, which can help identify and design new drug candidates.
1. Purpose of Docking
- To predict the binding mode of a ligand to its target.
- To estimate binding affinity using scoring functions.
- To aid virtual screening of large compound libraries.
- To guide lead optimization by showing structural fit and interaction patterns.
2. Types of Docking
a) Rigid Docking
- Assumes both ligand and protein remain rigid.
- Simplifies calculations but may miss conformational changes.
- Used when high-resolution structures are available.
b) Flexible Docking
- Allows flexibility in the ligand, protein, or both.
- More realistic but computationally intensive.
- Common in modern docking tools.
3. Steps in Docking Process
- Target Preparation
- Clean protein structure: remove water molecules (unless essential), add hydrogen atoms, assign charges.
- Define the active/binding site.
- Ligand Preparation
- Generate 3D structures, optimize geometry, assign protonation states.
- Docking Algorithm
- Searches for the best ligand pose in the active site.
- Examples: Genetic algorithms, Monte Carlo, Incremental construction.
- Scoring Function
- Evaluates poses based on predicted binding energy.
- Includes van der Waals forces, hydrogen bonding, electrostatics, and hydrophobic interactions.
- Post-Processing
- Analyze docking poses.
- Select the one with the best score and proper interactions.[3]
4. Common Docking Tools
- Auto Dock / Auto Dock Vina
- Molecular Operating Environment (MOE)
- Glide (Schrödinger)
- GOLD (Genetic Optimisation for Ligand Docking)
- Dock
5. Applications in Drug Design
- Virtual Screening – Rapidly testing thousands of molecules.
- Lead Optimization – Modifying chemical groups for better fit.
- Understanding Binding Mechanisms – Explaining activity differences.
- Predicting Drug Resistance – Studying mutation effects on binding.
6. Advantages
- Cost-effective compared to experimental methods.
- High-throughput screening possible.
- Provides atomic-level insights into interactions.
7. Limitations
- Accuracy depends on protein and ligand preparation.
- Scoring functions may not perfectly reflect real binding.
- Flexible proteins are harder to model accurately.
ADMET
ADMET stands for Absorption, Distribution, Metabolism, Excretion, and Toxicity — the key pharmacokinetic and safety properties that determine whether a drug candidate will work in the body as well as it does in the computer. It’s often evaluated in silico early in the design process to save time, money, and avoid failures in later animal/human studies.
1. Absorption (A)
- I. Goal: The compound should be absorbed efficiently into the bloodstream after administration.
- II. Key factors:
- III. Lipinski’s Rule of Five (MW ≤ 500, LogP ≤ 5, H-bond donors ≤ 5, H-bond acceptors ≤ 10).
- IV. Topological Polar Surface Area (TPSA) — <140 Ų preferred for oral drugs.
- V. Solubility — high enough to dissolve in GI fluids.
- VI. Permeability — predicted via models like Caco-2 permeability or PAMPA assays.[6]
- VII. Tools: SwissADME, ADMETlab, pkCSM.
2. Distribution (D)
- Goal: Once absorbed, the drug must reach the target tissue in effective concentration.
- Key factors:
- Volume of distribution (Vd) — indicates tissue penetration vs. plasma retention.
- Plasma protein binding (PPB) — high PPB can reduce free drug levels.
- Blood–Brain Barrier (BBB) penetration — essential for CNS drugs, undesirable for others.
- Transporter interactions — P-glycoprotein (P-gp) substrates may be pumped out, reducing efficacy.
3. Metabolism (M)
- Goal: The drug should be metabolically stable enough to remain active for its intended duration, but eventually metabolized to avoid accumulation.
- Key factors:
- Cytochrome P450 interactions — some drugs are rapidly metabolized, others inhibit P450 enzymes causing drug–drug interactions.
- Metabolite toxicity — some metabolites are harmful even if parent drug is safe.
- First-pass metabolism — significant for oral drugs; reduces bioavailability.
- Prediction tools: admetSAR, pkCSM, SMART Cyp.
4. Excretion (E)
- Goal: The body must be able to remove the drug or its metabolites efficiently.
- Key factors:
- Renal clearance — filtration and active secretion.
- Biliary excretion — drug excreted via bile into feces.
- Half-life (t½) — too short means frequent dosing; too long may cause accumulation/toxicity.
- Transporter effects — OATPs, MRPs, etc. affect excretion.
5. Toxicity (T)
- Goal: Avoid harmful effects at therapeutic doses.
- Key factors:
- Acute toxicity — LD?? prediction in animal models.
- Chronic toxicity — long-term effects on organs (liver, kidney, heart).
- Carcinogenicity — potential to cause cancer.
- Mutagenicity — DNA damage risk (e.g., Ames test prediction).
- Cardiotoxicity — hERG channel inhibition causing arrhythmia.
- Tools: Protox-II, admetSAR, Derek Nexus (commercial), Toxtree.
Typical In-Silico ADMET Workflow
- Input — structures of lead compounds from docking or virtual screening.
- ADMET prediction — using free or commercial tools.
- Filter — eliminate compounds with poor bioavailability or high predicted toxicity.
- Optimize — adjust functional groups to improve pharmacokinetics without losing activity.
- Re-test — predict again after modifications.
Why ADMET is Critical
- Around 50–60% of drug candidates fail in clinical trials due to ADMET issues, not lack of efficacy.
- Early prediction prevents late-stage expensive failures.
- Helps prioritize the most promising candidates for synthesis and biological testing.
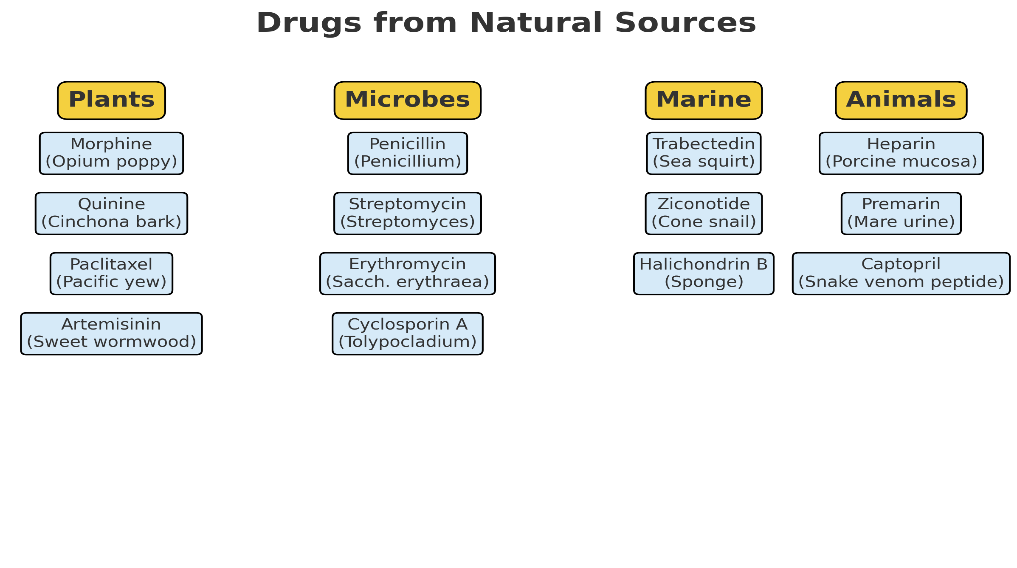
Sources of Natural Drugs
a. Plant-derived drugs
- Role: Plants produce secondary metabolites (alkaloids, terpenoids, phenolics, glycosides) with pharmacological activities.
- Examples:
- Morphine – analgesic (Papaver somniferum)
- Quinine – antimalarial (Cinchona bark)
- Paclitaxel (Taxol) – anticancer (Taxus brevifolia)
- Digoxin – cardiac glycoside (Digitalis lanata)
- Artemisinin – antimalarial (Artemisia annua)
b. Microbial-derived drugs
- Role: Microorganisms produce antibiotics and other bioactive molecules as part of their survival strategy.
- Examples:
- Penicillin – antibiotic (Penicillium notatum)
- Streptomycin – antibiotic (Streptomyces griseus)
- Erythromycin – antibiotic (Saccharopolyspora erythraea)
- Cyclosporin A – immunosuppressant (Tolypocladium inflatum)
c. Marine-derived drugs
- Role: Marine organisms produce unique metabolites due to extreme living conditions.
- Examples:
- Trabectedin – anticancer (Ecteinascidia turbinata, sea squirt)
- Ziconotide – analgesic (Conus magus, cone snail venom)
- Halichondrin B – anticancer lead (Halichondria okadai, sponge)
d. Animal-derived drugs
- Role: Some drugs are extracted from animal tissues or secretions.
- Examples:
- Heparin – anticoagulant (porcine intestinal mucosa)
- Premarin – estrogen replacement (pregnant mare’s urine)
- Captopril – ACE inhibitor inspired by snake venom peptides (Bothrops jararaca)
3. Advantages of Natural Drug Sources[6]
- Rich in chemical diversity
- Evolutionarily optimized for biological activity
- Can serve as templates for semisynthetic derivatives
- Often have novel mechanisms of action


 10.5281/zenodo.17204936
10.5281/zenodo.17204936
