Department of pharmaceutics, Dnyanvillas s College of Pharmacy, pune
Pharmacosomes represent an advanced and innovative drug delivery system designed to overcome the limitations associated with conventional dosage forms and vesicular carriers such as liposomes and noisome. They are amphiphilic complexes formed by the covalent conjugation of a drug molecule with a lipid, usually phospholipids. This unique structure enhances drug solubility, bioavailability, stability, and targeted delivery. Pharmacosomes have gained significant attention due to their ability to reduce dose-related toxicity, improve therapeutic efficacy, and provide controlled drug release. This review highlights the concept, preparation methods, characterization, advantages, limitations, and pharmaceutical applications of pharmacosomes, emphasizing their potential as an efficient drug delivery system.
Pharmacosomes are a new vesicular system for delivering drugs. In this system, medications are chemically connected to lipids. This creates colloidal dispersions that appear as very small vesicles, micelles, or hexagonal clusters. This lipid-based method improves bioavailability, stability, and accuracy of drug delivery. It also lowers toxicity and prevents leakage.[1]Pharmacosomes are lipid-conjugated vesicles where the drug itself plays a role in the structure of the vesicle. The name comes from the words "pharmakon" (meaning drug) and "soma" (meaning carrier). Pharmacosomes link harmaceuticals to phospholipids like phosphatidylcholine, often through active hydrogen atoms in the drug molecule. This linkage creates micellar, vesicular, or hexagonal aggregates. This is different from liposomes or niosomes, which simply encapsulate drugs. Since the drug-lipid combination stays intact during storage and transport, it allows for high drug loading, reaching up to 100% by weight, without the leakage problems that are common with traditional vesicles.[2]
Table 1: Comparison of conventional vesicular systems and pharmacosomes
|
Vesicular system |
Limitations |
Pharmacosomes |
|
Niosomes |
Drug leaching, time consuming, less stable |
More stable, more efficient. |
|
Liposomes |
Expensive, degradation by oxidation, sedimentation, leaching of drug. |
Cheaper to prepare, entrapment efficiency is independent of inclusion volume and drug bilayer interactions, covalent linkage prevents drug leakage, oxidation resistant and pure and natural phospholipids not needed. |
|
Transferosomes |
Expensive, oxidative degradation, lack of purity of natural phospholipids. |
Cheaper, oxidation resistant, pure and natural phospholipids not needed. |
Because of their amphiphilicity, which includes hydrophilic heads and hydrophobic tails, the structures can self-assemble into tiny particles, usually 50 to 200 nm, in water. By tackling issues like osmotic instability in liposomes, this design ensures stability across different pH levels and temperatures.
Pharmacosomes address some of the drawbacks of transferosomes, such as their vulnerability to oxidative breakdown and the purity of natural phospholipids. The prodrug gains both hydrophilic and lipophilic properties. It was found to reduce interfacial tension and exhibit mesomorphic behaviour at higher concentrations, similar to other components that form vesicles. The zwitterionic, amphiphilic, stoichiometric complexes of polyphenolic compounds and phospholipids are known as pharmacosomes. These lipid-based drug delivery systems are effectively designed because the colloidal mixtures of drugs with covalent, amphiphilic compounds allow the transfer of drugs through membranes, tissues, or cell walls within the body. They serve as a valuable method for achieving therapeutic goals such as controlled release and drug targeting.[4] [5]The surface and bulk interactions of lipids with the medication determine the requirement for vesicular pharmacosome formation. Any medication with or without a spacer chain, containing an active hydrogen atom (-COOH, -OH, -NH2, etc.), can be esterified to the lipid. This process produces a strong amphiphilic molecule.[2]
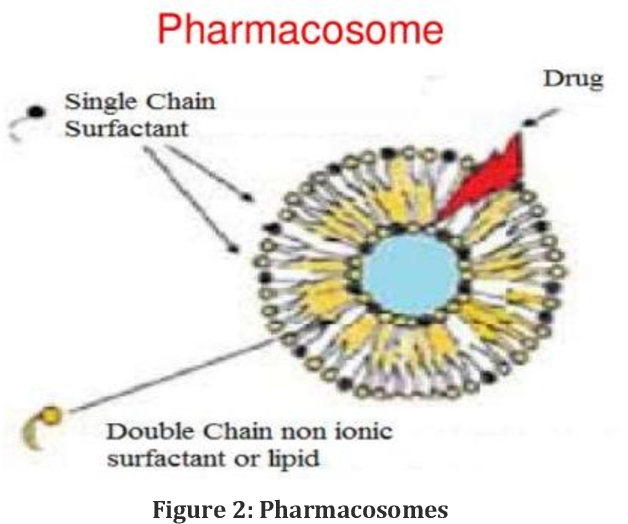
Figure no. 1
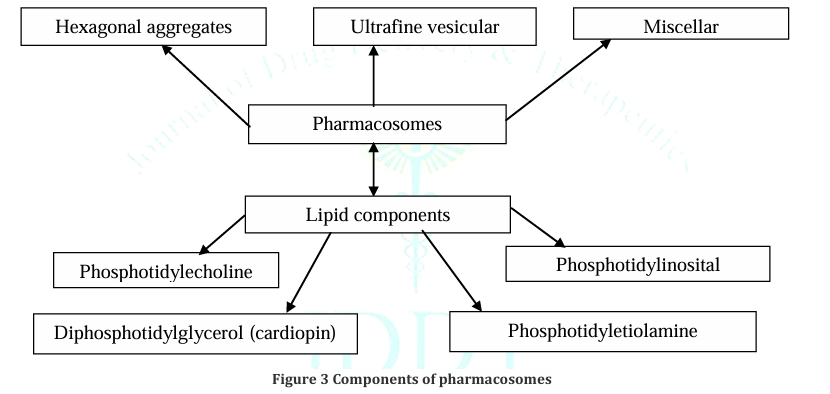
Figure no. 2
B .Pharmacosome Structures:
pharmacosomes can be classified as micellar, vesicular, or hexagonal based on the structure of the drug-lipid combination. These structures use covalent lipid binding to improve the solubility, stability, and bioavailability of drugs.[7]
Micellar pharmacosomes work well for drugs that do not dissolve easily in water. These are spherical structures with a hydrophobic core surrounded by a hydrophilic shell. They form when the drug-lipid conjugate is a single-chain amphiphile, which allows for leak-free solubilization in water.
Vesicular pharmacosomes have concentric lipid bilayers that surround an aqueous core. They resemble liposomes and work well for both water-soluble and fat-soluble medications. These tiny vesicles offer controlled release and high trapping efficiency. They come from two-chain amphiphilic conjugates.
Hexagonal pharmacosomes, commonly used for drugs that create rod-like conjugates, contain inverted cylindrical micelles arranged in a hexagonal pattern. Under electron microscopy, this type appears as tightly packed lipid cylinders and provides great membrane penetration.
C . Component of pharmacosomes :
Pharmacosome-compatible drugs must have active hydrogen atoms, like those in hydroxyl, carboxyl, or amino groups. This allows them to interact with lipids through esterification or amidation. Lipophilic medications, such as NSAIDs (like ibuprofen), cardiovascular drugs (like nifedipine), and anticancer agents, fit well because they can integrate directly into the lipid matrix with up to 100% loading efficiency. Adding hydrophilic medications can also boost solubility and bioavailability if they form stable complexes.
Any medication that has an active hydrogen atom (-COOH, -OH, -NH2, etc.)
An analytical-grade, intermediate polarity organic solvent is used to create pharmacosomes. High volatility and purity are important. In the chosen solvent, the drug and phospholipids must dissolve together. The solvent selection depends on the polarity of the drug and the lipid.
The main lipids are phospholipids. Phosphatidylcholine, also known as lecithin, is the most common because it is both amphiphilic and biocompatible. Other types include phosphoglycerides, sphingolipids, and sometimes free fatty acids or surfactants that help stabilize vesicular, micellar, or hexagonal forms.
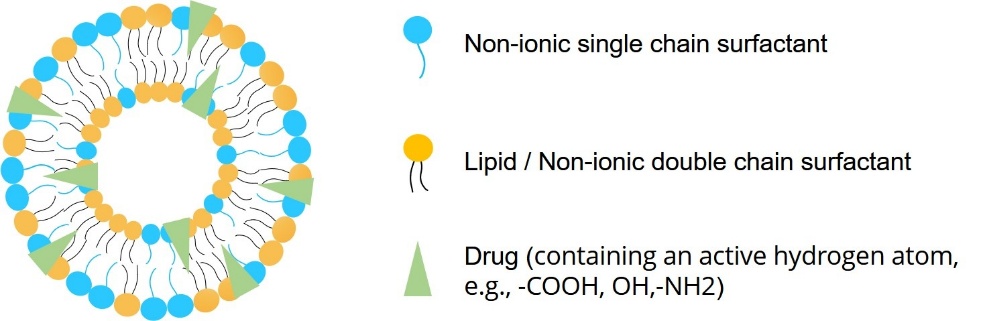
Figure no. 3
D. Function of Lipid Composition in Binding:
The formation and stability of drug-lipid conjugates inside pharmacosomes depend on the lipid composition. Phospholipids, like phosphatidylcholine, are commonly used due to their amphiphilic properties and compatibility with biological systems. A drug’s covalent bonding is helped by the presence of suitable functional groups, such as hydroxyl, amino, or carboxyl groups, in the lipid molecule. Changes in lipid composition affect membrane fluidity, molecular organization, and surface charge. These factors influence the binding strength, orientation, and stability of the drug-lipid complex. Therefore, choosing the right lipid can improve physicochemical stability, increase drug loading efficiency, and alter drug release characteristics.[8] [25] [26]
E. Phospholipid Fatty Acid Chains' Impact:
Pharmacosome characteristics depend heavily on the length and saturation of phospholipid fatty acid chains. Longer and saturated fatty acid chains can strengthen the drug-lipid connection. They also boost hydrophobic interactions, leading to more rigid and stable structures. However, this may slow down drug release. In contrast, shorter or unsaturated fatty acid chains increase membrane fluidity. This enhances flexibility and can improve drug incorporation and release rates. The type of fatty acid chain also affects particle size, aggregation behaviour, and how the particles perform in the body. Therefore, careful selection of fatty acid chain properties is essential to enhance the binding strength, stability, and effectiveness of pharmacosomes.[8] [9] [10]
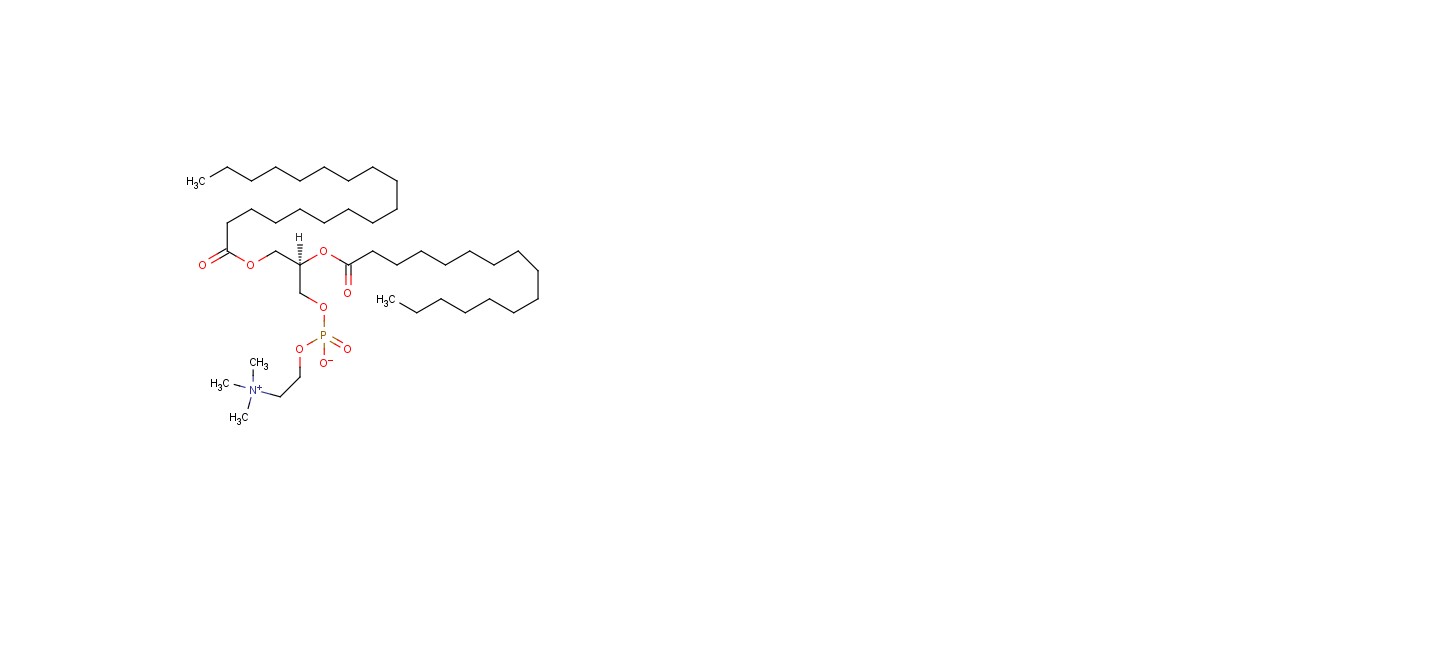
figure no. 4 Structure of phospholipids
F.Pharmacosome Physicochemical Characterization:[8] [17] [18]
1. Dimensions (nm):
The durability of pharmacosomes, their ability to load drugs, and their potential for cellular absorption all depend heavily on size. Smaller pharmacosomes may penetrate cells better and improve bioavailability.Pharmacosomes usually range in size from 70 to 150 nm. For the best stability and bioavailability, optimal formulations are often about 100 nm.
Particles that are smaller than 70 to 100 nm can enhance cellular absorption and penetrate biological barriers. However, they risk being quickly cleared by the reticuloendothelial system and may become unstable. This can reduce circulation time while improving targeted delivery.
2. Polydispersity Index (PDI):
The PDI indicates how the size of the pharmacosomes varies. A narrower size range suggests a more uniform population, as shown by a lower PDI.
3. Zeta Potential (mV):
The zeta potential represents the surface charge of the pharmacosomes. This charge affects their stability and how they interact with biological environments. If the zeta potential is negative, the pharmacosomes may be more stable and less likely to clump together
4. Intricate Determination:
FTIR spectroscopy confirms the formation of drug-lipid complexes by comparing the spectra of the complex, its individual components, and their combination, as well as identifying any peak shifts.
5. Surface Morphology:
SEM or TEM allows us to observe the size, shape, and surface structure of the vesicles. Lower lipid purity can lead to aggregates, while higher purity produces uniform spheres.
6. Efficiency of Entrapment
Free drug is separated by centrifugation or dialysis. Entrapment is measured by UV/HPLC, often reaching 70 to 90% in an ideal ratio.
7) Measurement of Crystal States
The X-ray diffraction technique helps determine the drug's crystal nature. You can adjust the tube voltage and tube current of the X-ray generator. Copper lines produce the radiation. The area under the curve of the X-ray diffraction pattern describes the specimen characteristics and shows the total integrated intensity of all reflection peaks.
8) Thermal Analysis
DSC detects endothermic peaks, phase transitions, and interactions, confirming complex stability and purity.
9) Drug release rate in vitro
The reverse dialysis bag method evaluates the in vitro drug release rate. This method involves placing the receiver phase outside the dialysis bag and introducing pharmacosomes inside. Each dialysis bag with the continuous phase is removed, and its contents are examined for drug release after being suspended in a vessel containing the donor phase and agitated at set intervals. One benefit of this method is the increased membrane surface area available for transport from the donor to the receptor compartment. Another benefit is the improved staffing efficiency due to fewer steps.
F. Mechanisms of Drug Binding
Drugs with active hydrogen atoms, such as -OH, -COOH, and -NH, serve as binding sites. They can form ester, amide, or thioester bonds when they react with lipid carboxyl or phosphate groups. These connections are influenced by spacer length and fatty acid chains to ensure optimal release after absorption. They offer strong stability against leakage and breakdown by enzymes.
Phosphatidylcholine is the main lipid that helps with vesicle formation because it is amphiphilic and biocompatible. The strength of the binding depends on the lipid content. Unsaturated fatty acids have spacers for steric chains that enhance fluidity and improve membrane contact for better penetration. On the other hand, saturated chains increase stiffness and stability
Affinity is influenced by factors such as temperature, pH, drug-lipid ratio, and functional group compatibility. Higher ratios can improve effectiveness by up to 90%, but they also raise the risk of aggregation. Improvement methods include control, solvent evaporation, or co-solvents. Detection uses HPLC to measure bound and free drug, and FTIR to confirm bonds.
G. Preparation method of Pharmacosome: [2] [11] [13]
1) The method of shaking hands
Combine the medication and lipid shell in a round-bottomed flask using the hand-shaking method. As the organic solvent evaporates at room temperature with a rotary vacuum evaporator, a thin layer of deposition forms on the walls. Hydrate the dry film with buffer and manually rotate it in one direction to create a vesicular suspension.
2) The method of ether injection
A set volume of ether dissolves the drug-lipid combination. The vesicles are created by slowly injecting this mixture into a hot buffer solution. The concentration influences the vesicle's properties, especially its shape. Depending on the amphiphilic condition, various structures can form, including round, cylindrical, disc, cubic, or hexagonal types.
3) The process of supercritical fluid
This technique is called "complex supercritical fluid solution enhanced dispersion." We achieve high supersaturation by sending the drug and lipid complex through the nozzle mixture chamber after they have been premixed in a supercritical carbon dioxide solution. Pharmacosomes form due to the rapid mixing of dispersion caused by the turbulent flow of carbon dioxide and solvent.
4) The method of solvent evaporation
First, we acidify the drug to make the active hydrogen available for complexation in the solvent evaporation method of pharmacosome preparation. After we extract it into chloroform, we recrystallize the drug acid. Then, we combine the drug acid and PC in different molar ratios to create the drug-PC complex.
5) The process of anhydrous co-solvent lyophilization
The drug and phospholipids dissolve in a solution of dimethyl sulfoxide containing glacial acetic acid. After mixing this solution until it is clear, it is freeze-dried overnight at condenser temperature. The resulting complex is stored at 4?C after being purged with nitrogen.
Table no. 2 Comparative studies different use in preparation of pharmacosomes
|
Method |
Vesicle Size |
Solvent Use |
Scalability |
Research Utility |
|
Thin Film Hydration |
Large |
Yes |
Low |
Basic research |
|
Ether Injection |
Small |
Yes |
Moderate |
Controlled delivery |
|
Solvent Evaporation |
Variable |
Yes |
High |
Industrial relevance |
|
Supercritical Fluid |
Nano |
No |
High |
Advanced nanomedicine |
|
anhydrous co-solvent lyophilization |
Variable |
Yes |
low |
Controlled delivery |
I.Advantages of pharmacosomes:[2] [11] [19]
1. Pharmacosomes perform better than other lipid-based drug delivery systems in many ways.
2. The drug-lipid complex is affected by the phase transition temperature, but the release rate stays the same because the drug is bonded to the lipid, which prevents leaching.
3. This system delivers drugs to specific sites, using hydrolysis as an enzymatic process to release the drug from the lipid polymer.
4. Factors like the spacer, lipid chain length, functional groups, and the size of the drug affect its metabolism during absorption.
5. They help lower therapy costs and work well for both hydrophilic and lipophilic drugs. Aqueous solutions of amphiphiles show aggregation based on their concentration.
6. With the drug and carrier linked together, the entrapment efficiency is high and consistent, and pharmacosomes aid in drug release through hydrolysis.
7. Pharmacosomes significantly improve the bioavailability of poorly soluble drugs while also reducing toxicity and side effects.
8. Unlike liposomes, pharmacosomes do not need to remove unentrapped drugs.
9. The use of pharmacosomes for drug delivery has improved the effectiveness of medications like bupranolol hydrochloride, pindolol maleate, acyclovir, and tax
J. Disadvantages of pharmacosomes: [2] [11] [20]
K. Application of Pharmacosomes: [2] [4] [6] [21]
L. Future Prospects of Pharmacosomes; [22] 23] [24]
Pharmacosomes are arising as a promising vesicular medicine delivery system, and ongoing exploration indicates a wide compass for their unborn development and clinical operation. The ensuing points punctuate the unborn prospects of pharmacosomes in pharmaceutical lores
1. Advancement in Targeted medicine Delivery
unborn exploration is anticipated to concentrate on face- modified pharmacosomes for point-specific and receptor- intermediated targeting, particularly for cancer, brain, and liver diseases. Ligand- attached pharmacosomes may enhance remedial efficacity while minimizing systemic toxin.
2. Advanced Bioavailability of inadequately Answerable medicines
With an adding number of recently discovered medicines flaunting poor waterless solubility, pharmacosomes offer a feasible platform to enhance solubility, permeability, and oral bioavailability, especially for BCS Class II and IV medicines.
3. Controlled and Sustained medicine Release Systems
unborn pharmacosomes phrasings may be optimized for controlled, sustained, and stimulants- responsive medicine release, using advanced lipid chemistry and polymer – lipid mongrel systems.
4. operation in Gene and Peptide medicine Delivery
Pharmacosomes may be explored for the delivery of peptides, proteins, and nucleic acids, which are generally unstable and inadequately absorbed. Lipid conjugation can cover these biomolecules from enzymatic declination.
5. Integration with Nanotechnology
The integration of pharmacosomes with nanotechnology could lead to nano- pharmacosomes with advanced cellular uptake, enhanced saturation, and better intracellular medicine delivery.
6. Development of Green and Scalable Manufacturing ways
unborn sweats will aim at developing cost-effective, scalable, and environmentally friendly manufacturing styles, easing artificial- scale product and commercialization.
7. Enhanced Stability and Shelf Life
Research on advanced lipid excipients and antioxidant objectification may significantly ameliorate storehouse stability and shelf life, prostrating one of the major limitations of pharmacosomes.
8. Personalized Medicine
Pharmacosomes can play an important part in substantiated and perfection drug, allowing customization of medicine – lipid conjugates grounded on case-specific remedial requirements.
9. Expanding remedial operations
unborn studies may expand the use of pharmacosomes in
• Oncology
• Neurodegenerative diseases
•Anti-HIV and antiviral remedy
• Transdermal and optical medicine delivery
• Vaccine delivery systems
10. Regulatory Acceptance and Clinical restatement
As further preclinical and clinical data come available, pharmacosomes are likely to gain nonsupervisory acceptance, paving the way for their restatement into marketable medicinal products.
CONCLUSION
Pharmacosomes are a new and promising concept within the pharmacy of medicine delivery systems, which possess many important advantages over typical lozenge-shaped medicine delivery systems, as well as other medicine delivery systems. The new approach of forming a covalent conjugate between the medicine to be delivered and the lipid itself is able to transcend the key hurdles of low soluble character, reduced bioavailability, medicine loss, and security. The potential for the unified delivery of controlled amounts of medicine, while at the same time efficiently improving remedial efficacy and reducing the levels of toxins, makes this approach highly attractive for the development of ultramodern medicine. Though there are challenges encountered in the large-scale product, cost, as well as comity of the medicine delivered
REFERENCES


Virashri Dhumal, Ajit Gholap, Atharav Muley, Pramod Ingale, Pharmacosomes: - A Novel Approach to Efficient Drug Delivery, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 2, 2707-2717. https://doi.org/10.5281/zenodo.18671915
 10.5281/zenodo.18671915
10.5281/zenodo.18671915
