1Department of chemistry, Yenepoya Institute of Arts, Science, Commerce and Management, Balmatta, Mangalore, 575002, India
2Yenepoya Research Centre, Yenepoya (Deemed to be University), Deralakatte, Mangalore, 575018, India
3Manipal College of Pharmaceutical Sciences, Manipal Academy of Higher Education, Manipal, 576104, India
Anthranilic acid is a benzenoid compound that has adjacent active important two reactive groups namely carboxylic acid group (-COOH) and amino group (-NH2). Consequently fascinating anthranilic acid derivatives have been prepared through both the active functional groups. The literature review reveals an experiential and holistic approach of various manufacturing procedures and skills used for the production of anthranilic acid analogues and anthranilic scaffold along with the biological activities of some of the promising derived anthranilic acid molecules. Pharmacology of anthranilic acid derivatives has been reported as alpha-glucosidase inhibitors, urease suppressors, keto-aldo reductase undecaprenyl pyrophosphate synthase (UppS) suppressors, mitogen activated protein kinase (MAPK), COX(Cyclooxygenase) inhibitors The analogues of anthranilic acid have been reported as an excellent pharmaceutical aids such as anticancer, antidiabetic, antiviral, anti-inflammatory, analgesic and antimicrobial agents. The present review unveils the importance of anthranilic scaffold as a beginning material for the development of potent drug for their various pharmacological activities.
Anthranilic acid is a precursor for the generation of therapeutic drugs in pharma industry. Analogues of Anthranilic acid is exhibited as pharmacophores for the rational development of medicinal drugs for administering the physiopathology and etiopathogenesis of numerous diseases. The derived products of anthranilic acid created a large compound library, which facilitates a holistic assessment of the structure activity relationship (SAR) analysis for the development of potent drug. Anthranilic acid plays a role as an intermediate in the production of azo dyes and saccharin in the synthetic industry. Esters of anthranic acid are used in perfumes for mimic jasmine and orange, pharmaceuticals (loop diuretics like furosemide), UV-absorber and corrosion inhibitors for metals and mold inhibitors in soy sauce. The research history includes N-phenyl anthranilic acid as an agent for inducing and studying renal papillary necrosis in the rat.(Hardy, 1970) and pharmacological properties of N-(3',4'dimethoxycinnamoyl) anthranilic acid, a new anti-atopic agent. (Azuma et al., 1976).
Anthranilic acid is a precursor for various available marketed drugs like furosemide (diuretic), betrixaban (anticoagulant), tranilast (antiallergic) and analgesic & anti-inflammatory fenamates. Nevertheless, anthranilic acid derivatives are used as potential anticancer, antiviral, antibacterial, antimicrobial, insecticidal, anti-inflammatory activities and other biological activities have been disclosed over the last three decades. Anthranilic acid is mainly a precursor in the biosynthesis of tryptophan and its derivatives. Tryptophan derivatives derived from anthranilic acid are cheap and efficient for the synthesis of various marketed available drugs like furosemide (diuretic), tranilast (antiallergic), betrixaban (anticoagulant) and analgesic & anti-inflammatory fenamates. Numerous anthranilic acid analogs with potential anticancer, antimicrobial, insecticidal, antiviral, anti-inflammatory activities and other biological activities have been disclosed over the last thirty years. (Nasr et al.,2022). The analogues of anthranilic acid have different functional head groups represented with –R group (R=Alkyl, aryl, any electronegative element group, etc) such as amides, amines and estersAnthranilic acid is a precursor for the generation of therapeutic drugs in pharma industry. Analogues of Anthranilic acid is exhibited as pharmacophores for the rational development of medicinal drugs for administering the physiopathology and etiopathogenesis of numerous diseases. The derived products of anthranilic acid created a large compound library, which facilitates a holistic assessment of the structure activity relationship (SAR) analysis for the development of potent drug. Anthranilic acid plays a role as an intermediate in the production of azo dyes and saccharin in the synthetic industry. Esters of anthranic acid are used in perfumes for mimic jasmine and orange, pharmaceuticals (loop diuretics like furosemide), UV-absorber and corrosion inhibitors for metals and mold inhibitors in soy sauce. The research history includes N-phenyl anthranilic acid as an agent for inducing and studying renal papillary necrosis in the rat. (Hardy, 1970) and pharmacological properties of N-(3',4'dimethoxycinnamoyl) anthranilic acid, a new anti-atopic agent. (Azuma et al., 1976).
Anthranilic acid is a precursor for various available marketed drugs like furosemide (diuretic), betrixaban (anticoagulant), tranilast (antiallergic) and analgesic & anti-inflammatory fenamates. Nevertheless, anthranilic acid derivatives are used as potential anticancer, antiviral, antibacterial, antimicrobial, insecticidal, anti-inflammatory activities and other biological activities have been disclosed over the last three decades. Anthranilic acid is mainly a precursor in the biosynthesis of tryptophan and its derivatives. Tryptophan derivatives derived from anthranilic acid are cheap and efficient for the synthesis of various marketed available drugs like furosemide (diuretic), tranilast (antiallergic), betrixaban (anticoagulant) and analgesic & anti-inflammatory fenamates. Numerous anthranilic acid analogs with potential anticancer, antimicrobial, insecticidal, antiviral, anti-inflammatory activities and other biological activities have been disclosed over the last thirty years. (Nasr et al.,2022). The analogues of anthranilic acid have different functional head groups represented with –R group (R=Alkyl, aryl, any electronegative element group, etc) such as amides, amines and esters.

Figure 1: Structure of anthranilic acid (AA) and analogues of AA.
In this review article the author’s interest is to showcase the classical and advanced synthetic approaches of the molecules derived from anthranilic acid and biological significance of anthranilic derivatives.
These derivatives are considered a cheap and efficient stating precursor for the production and synthesis of various marketed available drugs like furosemide (diuretic), tranilast (antiallergic), betrixaban (anticoagulant) and analgesic & anti-inflammatory etofenamates, analgesic (Glafenine, Floctafenine, Butylfluorofernate, Cyanthranipole) and chloranthraniprole (Insectside). Numerous anthranilic acid analogs with potential anticancer, antimicrobial, insecticidal, antiviral, anti-inflammatory activities and other biological activities have been disclosed over the last thirty years. (Nasr et al., 2022).

Figure 2: Structures of anthranilic acid derivatives showing analgesic, insecticide, diuretic and anticoagulant activity.

Figure 3: A precursor and intermediate molecule for the synthesis of oxazolobenzoxazine (16)

Figure 4: Structure of tricyclic benzoxazinone
Dehydration of anthranilic diacids (14) with acetic anhydride yielded the seven-membered 1,2-dihydro-4,1-benzoxazepine-3,5-dione (15), whereas treatment of the anthranilic diacids (14) with TFAA gave 3a-(trifluoromethyl)-5H-[1,3] oxazolo[3,2-a][3,1]benzoxazine-2,5(1H)-dione 16 (Figure 4)
Wang et al reported a synthetic method for the preparation of three Cu(II) complexes based on fluorinated AA derivatives [Cu(L1)(phen)].2H2O , [Cu(L2)(phen)] and [Cu(L3)(phen)] L1=4,5-difluoro-2-((2-hydroxybenzylidene)amino)benzoic acid, phen =1,10-phenanthroline) L2 =4,5- difluoro-2-(picolinamido)benzoic acid and (L3 =4-fluoro-2-(picolinamido)benzoic acid (Figure 5). The authors identified the interaction affinity between the synthesized complexes and calf-thymus DNA via using UV absorption, fluorescence spectroscopy and viscosity measurements. (Complex 17) marked the highest DNA binding affinity. Furthermore, they analysed the anticancer activity of the constructed compounds against A549 (human pulmonary carcinoma cells), Jurkat (human T lymphocyte cell line) and HepG-2 (human liver hepatocellular carcinoma cells) along with testing the antimicrobial activity using the agar-well diffusion method against E.coli (Gram-positive) and S.aureus (Gram-negative). The biological data revealed that (Complex 17) also acquired the highest activity against the three tested cancer cell lines (IC50= 1.42, 1.22, 7.09 µM for A549, HepG-2 and Jurkat, respectively) and bacteria. The authors concluded that the inhibition activity of complex 17 was attributed to two reasons. Firstly, the higher lipophilic character of complex 17 due to two fluoride atoms on the phenyl ring that could enhance its ability to cross the cell membrane. Secondly, complex 17 showed the highest DNA binding affinity, indicating that these complexes might target DNA to promote cell death. Fan.et.al (2019) discovered two cobalt complexes [Co2(L)(phen)(Ac)(PLC)], [Co3(HL)4(CH3O)2(H2O)2] and one zinc complex [Zn2(L)(phen)(Ac)(PLC)], (L = (2-carboxylato-5- (trifluoromethyl)phenyl) (3-hydroxy-4-methoxyphenoxy) amide, phen =1,10-phenanthroline, Ac = acetate, PLC =2-amino-4- trifluoromethylbenzoate) (Figure 5). The anticancer activity was carried out to cure human lung cancer cell (A549). And cervical cells. Nonetheless, the antimicrobial potency of the prepared complexes against E. coli (Gram-positive) and S. aureus (Gram-negative) using the agar-well diffusion method was also scanned. It was found that the zinc complex 22 was the most active molecule with a significant activity towards both A549 (IC50 =1.369µM) and Hela (IC50= 2.129?M). Antibacterial results also exhibited that complex 22 had the highest activity. The authors assumed that the high lipophilic fluorine- containing complex with Zn central atom exhibited better antitumor potency.

Figure 5: Structures of AA- metal complexes showing anticancer activities
The synthesis of series of 4-substituted benzenesulfonamides of AA was reported by Prachayasittikul and co-workers. The synthesized compounds were tested for cytotoxic, antifungal and antibacterial activity. The antiproliferative potency was examined against four cell lines (MOLT-3, HepG2, HuCCA-1 and A549) using etoposide and doxorubicin as reference drugs. The synthesized compounds showed cytotoxic activity against MOLT-3 cell line and compound 23 was the most potent one (IC50 = 15.71±0.70 µg/mL). In addition, the antibacterial activity was checked against eighteen strains of gram- negative and gram-positive bacteria (Figure 6). Unfortunately, none of the synthesized compounds possessed antibacterial activity. Nevertheless, all sulfonamides derivatives showed antifungal activity against C. albicans at micro molar concentration (4µg/mL) (26).

R=NO2, OCH3, CH3, Cl
Figure 6: Anthranilic acid containing potent anticancer agents.
In 2013, Shun Li et al reported the isolation and characterization of some AA derivatives from Penicillium paneum fungus (SD-44 ) found in deep sea sediment. The isolated compounds were tested for anticancer and antimicrobial activities. The cytotoxic activity was screened against two cell lines, Hela (human epithelial carcinoma cell line) and human colon cancer cell line (RKO) using fluorouracil as a reference drug. The biological activity revealed that compound (24) and (25) showed a potent anti-proliferative activity against RKO cell line (IC50 = 8.4 and 9.7 µM, respectively), whereas compound 26 exhibited cytotoxic activity against Hela cell line (IC 50=6.6 µM) which in both cases higher than the reference drug, fluorouracil (IC 50=25 µM against RKO cell line and 14.5 µM against Hela cell line) (Figure 6). Moreover, the antimicrobial activity was performed against two bacteria (Staphylococcus aureus and Escherichia coli) and three plant pathogenic fungi (Alternaria brassicae, Fusarium gramine arum, and Rhizoctonia cerealis), but unfortunately with no obvious activity [29].

Figure 7: Liu et al synthesised anticancer agents with anthranilic acid scaffold
Liu and coworkers (2013) prepared a novel series of anthranilamide derivatives and evaluated their anti-proliferative activity against two cell lines, human colon carcinoma cell line (HCT-116) and human breast adenocarcinoma cell line (MDA-MB-231). Compounds 27-28 exhibited a promising inhibition activity against the both cell lines and compound 27 (R=3-CF3) was the most potent one (IC50=14.6µM and 13.86µM against HTC-116 and MDAMB-231 respectively) (Figure 24). Flow cytometric analysis revealed that compound 27 (R=3-CF3) suppressed the proliferation of both cell lines through induction of apoptosis in a dose-dependent pattern and arrest G1 and S phase in the cell cycle [30]. El-Shafiey et al designed and prepared two AA derivatives, N-(2-carboxyphenyl) thipheneimine (HL2) and N-(2-carboxyphenyl) salicylideneimine (H2L1) and used them as a ligand for the synthesis of binary Ni (II), Co (II), Cd (II), Fe (III) and UO2 metal complexes and ternary pyridine metal complexes. The antibacterial activity is exhibited by these synthesized ligands, binary and ternary complexes were evaluated against Bacillus Subtilis, Escherichia Coli and Bacillus Cereus.
Joshi and co-workers synthesised a series of N-phenyl anthranilic acid derivatives and evaluated their anti-inflammatory activity using carrageenan induced rat paw edema method. The results exhibited that compounds 29 was the most active one with % inhibition 68.54% (Figure 8) [31].
Srivastava and co-worker represented the preparation and testing of anti-inflammatory and analgesic activity of series of 2-(4-oxo-2-phenylthiazolidin-3-yl)-5-(phenylazo)benzoic acids. The anti-inflammatory activity was evaluated with paw edema inhibition test and the reference drug was phenylbutazone. The analgesic activity was carried out using acetic acid in presence of reference drug, aspirin. Among the prepared compounds, the most potent compound in comparison with the standard drug at all doses tested was 30 (Figure 8) [32].

Figure 8: N-Aryl Anthanilic acid derivatives
Bala & co-workers designed and prepared a novel set of N-aryl AAs containing 1,3,4-oxadiazoles scaffold and evaluated their anti-inflammatory & analgesic activity in addition to molecular docking studies to survey their binding affinity to cyclooxygenase-2 enzyme. The anti-inflammatory activity was carried out using carrageenan-induced rat paw edema assay while the analgesic activity was performed using tail immersion method. The docking study indicated that compound 31 and 32 (Figure 9) formed a good interaction with COX-2 enzyme which explained the higher analgesic and anti-inflammatory activity of them. The authors believed that the good docking scores of compounds 31 and 32 were due to exchangeof the free –COOH which is responsible for gastric side effects with active 1,3,4-oxadiazole moiety [33]].

Figure 9: Anthranilic acid derivatives with 1, 3, 4-oxadiazole moiety
In 2013, Selley and co-workers studied in vivo analgesic & anti-inflammatory activity of N-(3,4-dimethoxycinnamonyl) anthranilic acid with collagen-induced assay,a mouse model of rhemattoid arthritis (Figure 10). They found that the compound 33 decreased the biological and histological intensity of arthritis, decreased the pain and eradicate the hyperalgesia. It also decreased cell activity in lymph node cell cultures and elevated serum levels of IL-10. The authors also studied the in vitro anti-inflammatory activity of compound 33 and founded that it caused suppression of IFN production and proliferation of both T and B lymphocytes [34].
![dimethoxyphenyl) prop-2-enoyl] amino} benzoic acid showing anti-inflammatory activity.png](https://www.ijpsjournal.com/uploads/createUrl/createUrl-20240730112441-5.png)
Figure 10: 2-{[(2E)-3-(3, 4-dimethoxyphenyl) prop-2-enoyl] amino} benzoic acid showing anti-inflammatory activity
Shi and coworkers reported the construction of a novel series of anthranilic diamides derivatives having arylisoxazoline moiety. In vitro anti-cancer potency was evaluated at micromolar, ?M level against cancer cell lines like human lung cancer cell line (NCIH 460), gastric cancer cell lines (SGC-7901 & BGC-823), breast epithelial adenocarcinoma epithelial cell (MCF-7) and hepatocellular liver carcinoma cell line (HepG2) using 5-flouro uracil as a reference drug. Compounds 34 and 35 showed a significant inhibition activity of the tested cell lines at 40 µg/mL and growth inhibition 59.2-76.3 %. Moreover, compound 36 exhibited a selective inhibition activity against human hepatocellular liver carcinoma cell type

Figure 11: Anthranilic acid derivatives with aryl isoxazoline moiety
Onnis and co-workers synthesized a series of esters of N-(2-(trifluoromethyl)-pyridin4-yl) AA derivatives based on the flufenamic acid scaffold. The prepared compounds were evaluated for anti-proliferative activity with almost 60 cell lines derived from leukemia, colon, lung, melanoma, CNS, renal, ovarian, prostate, and breast human cancers. Compounds 37 and 38 were the promising molecule against the majority o the tested cell lines at nanomolar concentrations (Figure 12). By using compare analysis, the authors believed that the most potent compounds exhibited their antiproliferative activity via COXdependent/independent mechanisms [35].

Figure 12: Anthranilic acid derivatives with aryl esters of N-(2-(trifluoromethyl)-pyridin4-yl)
Ullmann-Goldberg coupling reaction is a metal catalysed C-N bond formation coupling reaction in which copper catalysed coupling reaction is carried out for the preparation of N-aryl-AAs. In this reaction orthohalobenzoic acid reacts with aryl amine in presence of hot copper. Conversely, the reaction of AA derivatives with aryl halides using copper (metal, oxide, or salt) as a catalyst to produce N-arylAA analogs. To overcome the drastic reaction conditions (long reaction times and high temperatures), numerous modifications were proceeded on this coupling methodology utilizing a broader range of halobenzoic acid substrates, aryl amines and anilines along with using multiple types of bases and copper & copper complexes. In 2006, wolf et al reported an approach for the synthesis of N-alkyl and N-aryl AAs in high yields and without need of acid protection via regioselective copper-catalyzed amination of bromobenzoic acids with different aromatic and aliphatic amines.

Figure 13: N-aryl/alkyl anthranilic acid derivatives formed under regioselective copper-catalysed amination.
In 2003, Comdom et al also used ultrasonic irradiation for the construction of N-phenyl AAs 32 in high yields and short reaction time via coppercatalyzed Ullmann-Goldberg coupling reaction using water as a solvent

X=Cl, Br
R, R1=F,Cl,Br, OCH3, NO2, OH, CN, COOH, alkyl, aryl
Figure 14: Construction of N-phenyl Anthranilic acid derivatives
Culf et al demonstrated a novel one-pot reaction attempt for the construction of AA derivatives 50. This SNAr reaction involved the reaction of substituted 2-fluorobenzoic acid 48 with diisopropylcarbodiimide (DIC) 49 in solvent mixture of 1-Propanol (n-PrOH) and N-Methyl-2-pyrrolidone (NMP) (Figure 15). This methodology is simple, metal free, regiospecific and tolerable for 2-florobenzoic acid with many electrons withdrawing substituents

Figure 15: Reaction of AA derivatives with diisopropylcarbodiimide
The substituted anthranilic acid can be diazotised like amines, rather zwitter ionic inner salt formed in non-aqueous media. The benzenediazonium carboxylate synthesised in alcohol with isoamyl nitrite, can also be isolated and is stable below -700C. On warming to 40-600C structure of anthranilic acid fragmented to N2 gas, CO2 and benzyne and can be used as a source of the highly reactive compound. The formation of a closed ring structure, a corresponding 1,2,3-benzotriazine-4-ones has been discussed. There is a published review on the use of arynes in the preparation covering the years 1990-2002 with refrence to former reviews.

Figure 16: Formation of benzenediazonium carboxylate and benzyne
N, N-dimethylaniline and anthranilic acid diazotised to form diazodye and methyl red like amines. Conversion of anthranilic acid into corresponding 2-nitrobenzoic acids is an example for N-oxidation. Oxidation of anthranilic acid to give 2-nitrobenzoic acid can be carried out using superoxide radicals generated from hydrogen peroxide on soid titanium catalyst. Pperoxyacetic acid or peroxymonosulfuric acid (Caro’s acid) is an oxidising agent used for the oxidation of anthanilic acid into 2-nitrobezoic acid. Compound 57 can also be synthesised by oxidation of N-hydroxy-anthranilic acid and combination of AA and sodium perborate. Anthranilic acid can be reduced to 2-aminophenylmethanol using sodium amalgam (Na-Hg), lthium aluminium hydride or samarium salts (At room temperature). Reductions of esters to alcohols have been reported with sodiumborohydride or ZnCl2/NaBH4. Birch reduction process can also be used to reduce anthranilic acid.

Figure 17: Various forms of Anthranilic acid derivatives
2-[(trifluoroacetyl)amino] benzoic acid (59, Figure 18) prepared by treatment of anthranilic acid with trifluoroactic acid (TFAA) [36] [37], could be activated with dicyclohexylcarbodiimide (DCC) and coupled with alcohols to prepare intermediates for natural product synthesis [38]. Subsequently deprotection of trifluoroacetyl group from 2-[(trifluoroacetyl)amino] benzoic acid processed with sodiumborohydride. N-acetyl anthranilic acid derivatives are obtained after the reaction of sodium salt of anthranilic acid with acetic anhydride followed by acidification of HCl [39]. N-protection in anthranilic acid carried with Boc group, either directly [40], or through reductive condition from 2-azidobenzoic acid [41]. N-Boc-anthranilic acid on reaction with Grignard reagents produce the corresponding 2-aminobenzoketones with optimum yield [42]. N-Tosyl anthranilic acid has been synthesised and used in Friedel crafts acylation of its acid chloride [43]. Deprotection needed strong acidic condition (hot conc. H2SO4) [44].
O-protection of anthranilic acid is essential to esters of anthranilic acid as a precursor e.g., methyl anthranilate. Potentially the easily prepared cyanomethylester (60) can also be used. . Removal of protective group performed by treatment [45] or simply through basic hydrolysis [39]. Boron compound have been used to protect both amino and carboxyl group of anthranilic acid [46].

Figure 18: Formation of anthranilate, cyanoanthranilate and boroanthranilate
Medicinal Chemistry of Anthranilic Acid: The non-steroidal analgesic and anti-inflammatory drugs having anthranilic acid derivatives such as glaenine (62), Fenamates, Etofenamate (63) and Flufenamic acid (64), (Fig. 19). The pharmacology of the synthesised compounds have reviewed [47, 48]. Hypersensitivity to these non-steroidal anti-inflammatory drugs has also been recorded [49]. The 3-substituted anthranilate hydroxamic acids 65 (Fig. 20), synhesised from 2, have been shown to inhibit matrix metalloproteinase’s (MMPs), enzymes that play a role in the remodelling in degradation of extracellular matrix proteins and have been implicated in the etiology of several diseases such as rheumatoid arthritis and cancer [50]. The amides 66 have been shown to be good CCK1 (cholecystokinin, an endocrine and nervous system cellular messenger) receptor antagonists (R=carboxy, indolyl/naphtyl) [51]. Anthranilic derivatives with a similar structure i.e. 67 (Fig. 4) have been exhibited as effective inhibitors with protein-tyrosine phosphatises, i.e., a category of enzyme in which attractive target accomplished such as diseases like diabetes, cancer and inflammation [52, 53]. Some of the anthranilic acid used as building block for peptidomimetic drugs. Even AA can act as protease inhibitor [54]. There is a drug Tranilast, an avenanthramide, was originally developed as an anti-allergy drug but it has been shown to inhibit angiogenesis [55], which is an important phenomenon in cancer and other degenerative diseases. Recently AAs with pyridine analogues like Flufenamic acid (64) have been reported as anticancer agents [56].

Figure 19: NSAID’sand analgesic drugs like Glafenine, Etofenamate and Flufenamic respectively.
![Anticancer agents such as N-hydroxy-2-{[(4-methoxyphenyl) sulfonyl] amino}-3-methylbenzamide derivative, {[2-(amino) benzoyl] amino} acetic acid derivative and 2-[(carboxycarbonyl)amino] benzoic acid derivatives respectively.png](https://www.ijpsjournal.com/uploads/createUrl/createUrl-20240730111636-15.png)
Figure 20: Anticancer agents such as N-hydroxy-2-{[(4-methoxyphenyl) sulfonyl] amino}-3-methylbenzamide derivative, {[2-(amino) benzoyl] amino} acetic acid derivative and 2-[(carboxycarbonyl)amino] benzoic acid derivatives respectively
Synthesis of five membered heterocycles from anthranilic acids
N-alkyl substituted AAs produce many heterocyles on self-condensation. This chemistry has its origin in Carl Heumann’s classic indigo synthesis from N-carboxymethyl-AA (14)[35]. The deep blue indigo product obtained on heating with K/Na hydroxide directly. Later O, N-diacetyl indoxyl (69) were synthesised by treating with acetic anhydride/ sodium acetate. The procedure was given in a German patent [58, 59], with two experimental details [60, 61]. A experimental procedure is outlined in other publications [62, 63]. The formation of mixed anhydrides of the starting materials with acetic acid, followed by a base induced ring closure and loss of CO2 [64], with a process like Perkin reaction. The reaction was carried out with variously ring substituted derivatives of 8 [48, 64-67], along with side chain substitution [68]. Compound 33 could be O-deacetylated with sodium thiosulfite (i.e. Na2S2O5 dissolved in water) to 34[63, 67], and transformation from N-deacetylated to free indoxyl (35) under basic aqueous oxygenated conditions [66]. Under oxidative condition, the indoxyl dimerises and forms indigo [62]. The followed mechanism is annihilation of two radicals of indoxyl [68]. The equilibrium constants of keto-enol transformations of indoxyl have been determined (and its O and S analogues) [159].


Table: Substituent’s (R1, R2 & R3) in Condensations of 2-substituted indoxyls
In spite of the nature of R1 (Figure 2) only metal alkoxides haave been used as the bases with long time heating when R1=H. But when R1=alkyl the reaction can be carried out at room temperature, where as R1=aryl the reaction can be carried out at 00C at room temperature.

Figure 22 Condensation yielding 2-substituted indoxyls derivatives (73).
The chemistry indoxyl has been reported [70], but the following some examples are extended reactions of indoxyls. A procedure for hydrolysis/decarboxylation followed by condensation with amines [71], has been used to make the compounds 38 (Figure 23) [72]. Indoxylic derivative (74), the possible intermediate in the Heumann indigo synthesis, that is at the maximum conveniently prepared by hydrolysis of the ethyl ester [73], has been made to react with 2-aminobenzaldehyde to form [3, 2-b] quinoline by decarboxylation condensation [74]. Indoxyl (or N-acetyl indoxyl, or O, N-diacetyl indoxyl) has been condensed with isatin (in its ring opened form) in basic aqueous solution to give the acid [74-77].

Figure 23: Decarboxylative amination for 1-phenyl-3-(piperazin-1-yl)-1H-indole derivatives, (X, Y = H or F).
The indoxyl derivative (76) has been cyclised to indoloindol (77) by heating with acetic acid (Figure 24) [78].
![Synthesis of 1, 4-dihydropyrrolo [3, 2-b] benzopyrrole derivatives.png](https://www.ijpsjournal.com/uploads/createUrl/createUrl-20240730111636-10.png)
Figure 24: Synthesis of 1, 4-dihydropyrrolo [3, 2-b] benzopyrrole derivatives
On treatment of anthranilic diacids 78 with Vilsmeyer reagent, followed by heating produces indoles 79 in varying yields (Figure 25) [79]. The reaction can also be used to other diacids for the preparation of non-benzofused heterocycles.

Figure 25: Synthesis of Indole, Benzofuran and Benzothiophene derivatives via cyclisation by Vilsmeyer reagent (X=S, O, NH and R=H, Me, Br, I).
In case diazotised anthranilic acid is allowed to react with Na2S2 (prepared from Na2S and S8), dithiosalicylic acid (81) has been formed (Figure 26)[80]. It can easily be reduced to thiosalicylic acid (85)[80], and afterwards reacted with chloroacetic acid under aqueous basic conditions to produce the diacid under aqueous basic conditions to give the diacid 83 [81]. The prior report demonstrated the direct conversion of 48 to the diacid 83 with reducing agent (sodium dithionite). Cyclisation by Vilsmeier reagent reactions leading to benzothiophenes and thioindigo. [82], but some preparative procedures carry out reactions without the addition of a reducing agent [83, 84]. Benzo [1,2] dithiol-3-one (84) can be synthesised from 81 [85] or from 85 [81,85,86]. The diacid 80 is made to react with acetic anhydride to give benzothiophene {84), which could be deacetylated to benzothiophene derivative (84). Compound 83 is directly converted to 86 [87]. Like its nitrogen counterpart indoxyl, 86 can be dimerized to the corresponding indigoid compound, in this case thioindigo (88). A mild procedure has been developed with hydrogen peroxide in acetic acid [88]. The dihydrobenzothiophene 86 has been synthesised from 83, and further converted to the thioindigos 88 which is used to synthesise photo activated molecular tweezers [83,84]. Benzothiophenes have been prepared from 83 (Figure 25) 1, 2-Benzothiazol-3(2H)-ones, which exhibit antimicrobial activities, have also been prepared from 81, a review has been reported [89, 90]. An ecofriendly process has been developed to prepare derivatives of 1, 2-Benzothiazol-3(2H)-ones from esters of 81 [91]. There are many syntheses of oxidised derivatives of the sweetener saccharin from anthranilic acid. A recent example utilizes diazotisation of methyl anthranilates [92].

Figure 26: Synthesis of benzothiophene derivatives
2, 1-Benzisoxazole (90: R1=R2=H in Figure 27), also known as anthranil, having special valence tautomerism and aromatic properties [91]. There is a preparation in the literature based on oxidation of methyl anthranilate derivative [92] The treatment of isatoic anhydride with chiral amino alcohols give the anthranilic amides (92) (Figure 28) [93]. These can be ring closed through the compounds (93) to the oxazoles 94 which were utilised in the synthesis of tridentate ligands for asymmetric organometallic catalysis.

Figure 27: Synthesis of a 2,1-benzisoxazole (90: R=Me, R1=COOMe).

Figure 28: Synthesis of 2-(4,5-dihydro-1,3-oxazol-2-yl)aniline (R=H, alkyl & halogens)
Benzimidazoles of anthranilic acid reacted with o-phenylenediamine in very hot polyphosphoric acid [94, 95], or under basic conditions [96], to form the benzimidazole derivative 97 (Figure 29).

Figure 29: Synthesis 2-(1H-benzimidazol-2-yl) aniline
The 2,3-diaminobenzoic acid 98 has undergone cyclisation to give the benzimidazole 99 on treatment with formic acid [97, 98], or to give benzimidazolone 100 on treatment with phosgene (Figure 30) [99]
![Synthesis of different forms of benzimidazole on Curtius rearrangement. (R=H [105], COPh [100], COOMe [101], C (CH3)3 [102])..png](https://www.ijpsjournal.com/uploads/createUrl/createUrl-20240730111636-4.png)
Figure 30: Synthesis of different forms of benzimidazole on Curtius rearrangement. (R=H [105], COPh [100], COOMe [101], C (CH3)3 [102]).

Figure 31: Synthesis of 1, 3-dihydro-2H-benzimidazol-2-one derivatives
Many benzimidazol-2-ones (102) were prepared on heating the anthraniloyl azides 101)(Figure 31). The products were formed using Curtius arrangement followed by internal capture of the intermediate isocyanate. The precursor 101 are obtained by diazotisaton of the corresponding Anthranilic hydrazides [103], or by ring opening of isatoic anhydride or phthalic anhydride by azide ion [105].
![Synthesis of 1,3-dihydro-2H-benzimidazol-2-one derivatives by Lossen rearrangement (R1= Et or Me, R2 = H, Me or Et, X and Y = Br, Cl or H) [106]. .png](https://www.ijpsjournal.com/uploads/createUrl/createUrl-20240730111636-2.png)
Figure 32: Synthesis of 1,3-dihydro-2H-benzimidazol-2-one derivatives by Lossen rearrangement (R1= Et or Me, R2 = H, Me or Et, X and Y = Br, Cl or H) [106].
The benzimidazol-2-ones were prepared with high yields (71-99 %) on heating of hydroxamic acids of AA in formamide [106]. This is a modified Lossen rearrangement which appears to give reaction directly from the corresponding hydroxamic acid, circumventing the need to first O-acylate the starting material. The derivatives of hydroxamic acids were synthesised by reaction of appropriately substituted anthranilic ester with hydroxylamine hydrochloride. The sydnones (107) were prepared by dehydration of N-nitroso derivatives (106) (Figure 33).

Figure 33: Synthesis of sydnones

Table: List of literature with substituent’s of sydnone derivatives
Synthesis of six membered rings Rutacea family plants possess Acridine alkaloids derived from anthranilic acid [107]. The extraction, isolation, characterisation and synthesis of alkaloids are periodically reviewed [108]. There are enormous syntheses of acridones and acridines from N-aryl anthranilic acid in the literature. The two main schemes given in Figure 34. Acridone (109).
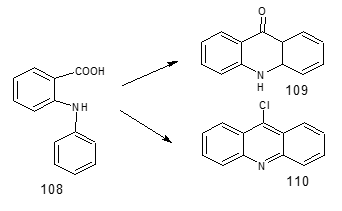
Figure 34: Synthesis of acridines
An illustration of the first type reaction is cyclisation of the AA derivative (111) (available by Jourdan-Ullman-Goldberg reaction of 2-halobenzoic acid and AA) to the acridone 112, followed by reduction/oxidation to the fully aromatic acridine 113 (Figure 34) [109]. The reaction of 111 with Trifluoroacetic anhydride followed by heating gives ring closure to 112[110].
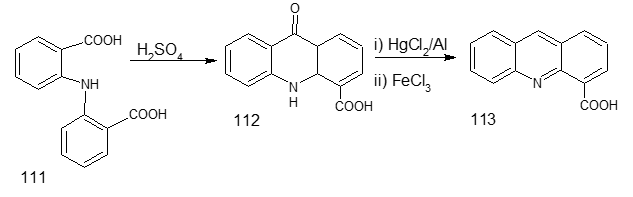
Figure 35: Synthesis of acridine-4-carboxylic acid
Synthesis of anti-Herpes simplex acridones are the reactions of anthranilic acid with resorcinols at high temperatures are prepared to yield the acridones 116, which is similar to naturally occurring acridones (R1 -R4) are combined with different substituents like H, Cl, Me and OMe, R5=OH, OMe or Me)(Figure 36)[111]. The former by initial N-thioacylation of anthranilic acid with thiobenzoylthioacetic acid, followed by ring closure in acetic anhydride [112]. The later compound, 77, was directly prepared by heating of anthranilic acid in thioacetic acid [113].

Figure 36: Synthesis of highly substituted acridines
9-disubstituted acridines (118) are formed by the treatment of various substituted esters of N-phenyl anthranilic acid with Grignard reagents (Figure 37), followed by ring closure [117].

Figure 37: Synthesis of 9, 10-dihydroacridine derivatives (Y=H, Cl, F, OCH3, alkyl and NO2)
Reaction of ammonium isothiocyanate (NCS)NH4 with anthranilic acid gives the thiourea derivative [118], but reaction with benzoyl isothiocyanate gives 120 (Figure 38) [119, 120]. The later product can be cyclised under acidic conditions to give benzothiazine 121, which also hydrolysed to 122, and later transformed by Dimroth rearrangement to give thioxoquinazolinone 123 (under basic or acidic conditions) [121]. The transformations were carried out with several ring substituted derivatives of 120. In the similar way the reactions of anthranilic acid with phenyl isothiocyanates were found to yield benzothiazines or thioxoquinazolinones depending on the substitution pattern in the starting marterial [122].

Figure 38: Synthesis of 2-thioxo-2, 3-dihydroquinazolin-4(1H)-one
The following scheme for the synthesis of 1, 2, 3-benzotriazin-4(3H)-one derivatives (formation of benzotriazinones by diazotisation of methyl antthranilate) There was a problem on traditional synthesis of benzotriazinones 127is to prepare the substituted anthranilamide The problem can be solved by addition of the appropriate amine (Figure 39) [123]

Figure 39: Synthesis of 1, 2, 3-benzotriazin-4(3H)-one derivatives(R=alkyl or aryl)
A holistic synthesis of benzotriazine (130) as an additive in peptide couplings have been developed using the reaction of 128 with TMS-protected hydroxylamine to give 129, followed by nitrosation (yielding 36-68?pending on substitution on 129) (Figure 40) [124]. Thermolytic [125,126] and photolytic [127] studies on 1, 2, 3-benzotriazine-4-ones have been published.

Figure 40: Synthesis of 3-hydroxy-1, 2, 3-benzotriazin-4(3H)-one derivative (X=Y= F, Cl, NO2, alkyl, OCH3 and H)
When AA is condensed with an aldehyde (R= alkyl or aryl) the following substituted 1, 2-dihydro-4H-3, 1-benzoxazin-4-one (133) have been formed through intramolecular nucleophilic attack and a proton shift (Figure 41). The final product formed is much more stable than the intermediate imine and cannot be isolated normally. The reactivity is simple to prepare such that cyanide opens the ring by addition to the 2-position (under basic conditions the salt of the cyanomethylated anthranilic acid obtained), and the same chemistry has been used to prepare functionalised starting materials for the synthesis of indoxyls (Figure 42)

Figure 41: Synthesis of substituted 1, 2-dihydro-4H-3, 1-benzoxazin-4-one
The derivatives of (4H)-3-benzoxazine-4-ones have been formed by intramolecular nucleophilic attack in N-acetylated anthranilic acid, but it is not variable process due to low electrophilicity of amide. Rather the reaction is induced by formation of a mixed anhydride of the starting material, usually by heating in acetic anhydride. The amide functions as nucleophile in its iminol form. Which cannot be normally isolated eliminates simpler water molecule to form the product. The chemistry of the resulted compounds sometimes called ‘acylanthranils’ has been reviewed (covering the period 1965-1998) [128]. A suitable way to synthesise (4H)-3-benzoxazine-4-one’s derivatives, i.e., inhibitors of C1 serine protease, is to treat with anthranilic acid and benzoyl chloride in pyridine [129]. Similar compounds have been isolated from oats (Avena sativum) [130]. The usage of cyanide ring opening (X=H or F). The alkylation procedure has been published in elsewhere [131].
![Synthesis of derivatives of 2-[(2-methoxy-2-oxoethyl)(phenyl)amino]benzoic acid(X=H, F, Cl, and alkyl).png](https://www.ijpsjournal.com/uploads/createUrl/createUrl-20240730111439-11.png)
Figure 42: Synthesis of derivatives of 2-[(2-methoxy-2-oxoethyl)(phenyl)amino]benzoic acid(X=H, F, Cl, and alkyl)
The method in which N-acyl AA is reacted with anilines to give 2, 3-disubstituted-4-quinazolinones catalysed by PCl3 [132], has been reproduced and the conditions optimised [133]

Figure 43: Synthesis of quinazolin-4(3H)-one and quinoline derivatives (R1=R2=substituted alkyl or aryl)
The Niementowski quinoline synthesis in which 3-hydroxy-2,3-dihydroquinolin-4(1H)-one derivatives have been synthesized from anthranilic acid and acyl derivatives in presence of strong base in turn treating with polyphosphoric acid (PPA) yielding to quinoline derivative (147).

Figure 44: Synthesis of quinolin-4(1H)-one derivatives (R1=alkyl or aryl)
Synthesis of quinolinones 151 from N-hydroxy-2-nitroethanimine and anthranilc acid as prime moiety in which R group can be varied with different substituents. (R1=SMe and R2=COAr [134], or R1=NHPh, R2=NO2 [135])

Figure 45: Synthesis of 3 & 4 substituted quinoline moiety.
Synthesis of quinolinones 154 from methyl prop-1-en-1-yl sulfide and AA in presence of cyclising reagent. (R1=SMe and R2=COAr [141], or R1=NHPh, R2=NO2 [135])

Figure 46: Synthesis of 2 & 3 substituted quinoline moiety.
Synthesis of seven membered rings
There are many cyclic seven membered alkaloids derived from anthranilic acid that appear in nature [136]. Among them some are of the type 1,4-benzodiazepine-2,5-diones (Figure 47) or straightaway derived from such compounds. It means that they are biosynthetically built from anthranilic acid and amino acids. DNA-binding anthramycin derivatives and anthranilic acid derivatives are the starting materials in the most published report. The chemistry has been reviewed [137]. The most usual way of making compounds type has been to allow an amino acid (or esters thereof) to attack isatoic anhydride [138,139]. Heating of anthranilic acid made that induces cyclisation to (158, Figure 47). Instead the amide could be isolated and the ring is closed by heating with acetic anhydride. [140]. The stereochemistry of the starting material retained in the product. Another approach utilizes DCC mediated coupling of NBoc-AA and oxazolidine-2,5-diones (N-carboxyanhydrides) derived from amino acids [141]

Figure 47: Synthesis of 3 substituted- 3, 4-dihydro-1H-1, 4-benzodiazepine-2, 5-Dione (R1=R2=H or alkyl)
The anthranilic acid derivatives 159, with EWG (electron withdrawing groups) attached to the nitrogen of anthranilic acid have been cyclised to the 1,4-benzodiazepine-3,5-diones 160 on reacting with chloroformates and triethylamine (Figure 48) [69]. In the cases where X=NO the nitrogen could be deprotected.
![Synthesis of N-substituted-1H-1,4-benzodiazepine-3,5(2H,4H)-dione (X=COOEt, Ac, COOBn, COOMe or NO, R=H, Et or Ph} [69].png](https://www.ijpsjournal.com/uploads/createUrl/createUrl-20240730111439-5.png)
Figure 48: Synthesis of N-substituted-1H-1,4-benzodiazepine-3,5(2H,4H)-dione (X=COOEt, Ac, COOBn, COOMe or NO, R=H, Et or Ph} [69]
Preparation of benzotriazepinones (X= H, Me or Cl) in which benzotriazepinone 164 has been synthesised from anthranilic acid on reaction with a nitrilimine, occurred in situ from 162, followed by ring closure with peptide coupling reagent (Figure 49) [142]. The structure 164 was later confirmed by X-ray crystallography [143].
![Synthesis of 2-acetyl-4-(4-methylphenyl)-3,4-dihydro-5H-1,3,4-benzotriazepin-5-one and 1,3a,4,5-tetrahydro-6H-pyrrolo[1,2-a][1,4]benzodiazepin-6-one derivatives (R=R1=R2=alkyl, F, Cl).png](https://www.ijpsjournal.com/uploads/createUrl/createUrl-20240730111439-4.png)
Figure 49: Synthesis of 2-acetyl-4-(4-methylphenyl)-3,4-dihydro-5H-1,3,4-benzotriazepin-5-one and 1,3a,4,5-tetrahydro-6H-pyrrolo[1,2-a][1,4]benzodiazepin-6-one derivatives (R=R1=R2=alkyl, F, Cl)
Synthesis of 4,1-benzoxazepine-5-ones 169 in which compound 168 treated with polyphosphoric acid (R=Ph or Me) [292]

Figure 50: Synthesis of 4,1-benzoxazepin-5(1H)-one derivative (R=alkyl or aryl)
Macromolecules
Reaction of methyl anthranilate (170) with a strong base produces a dimerised 8-membered ring 171 with good yield. (Figure 51) [144]. The same product can also be obtained by intramolecular peptide coupling of anthraniloylanthranilic acid (173) [144], available by reaction of isatoic anhydride (5) with AA. The latter route lends itself to preparation of non-symmetric derivatives of 171 [144,145].
By coupling of N-alkylsubstituted derivatives of 173 with yet another AA unit the trianthranilides 175 have been prepared. The resulted could also be cyclised to the macrocycles 176 (Figure 52) [144].
The anthranilic dimmers (174) have produced the macrocyccles (178). Which were obtained by reaction of isatoic anhydride with diamines (Figure 53) [146].
![Synthesis of dibenzo [1,5] diazocine-6,12(5H,11H)-dione (R=alkyl or aryl).png](https://www.ijpsjournal.com/uploads/createUrl/createUrl-20240730111439-2.png)
Figure 51: Synthesis of dibenzo [1,5] diazocine-6,12(5H,11H)-dione (R=alkyl or aryl)

Figure 52: Synthesis of (3Z,7Z,11Z)-1,5,9-triazacyclododeca-3,7,11-triene-2,6,10-trione derivatives
![Synthesis of 16,17-dihydrodibenzo[e,l][1,3,7,11]tetraazacyclotetradecine-6,8,14,18(5H,7H,9H,15H)-tetrone derivatives.png](https://www.ijpsjournal.com/uploads/createUrl/createUrl-20240730111439-0.png)
Figure 53: Synthesis of 16,17-dihydrodibenzo[e,l][1,3,7,11]tetraazacyclotetradecine-6,8,14,18(5H,7H,9H,15H)-tetrone derivatives
CONCLUSIONS
The anthranilic acid analogues have shown vital role in the treatment of lethal diseases. The analogues of anthranilic acid have intended to control carcinogenic pathways, metabolic complications related to diabetes, cutting edge antiviral agents and physiologically tolerable anti-inflammatory compounds.
REFERENCE



Chandrashekhara Kumar B*, Arun A Bhagwath, Chandrashekar K S, Pharmaceutical chemistry of anthranilic acid derivatives: A brief review, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 7, 2143-2174. https://doi.org/10.5281/zenodo.13131906
 10.5281/zenodo.13131906
10.5281/zenodo.13131906
