Shiva Trust's Godavari College of Pharmacy.
Cancer therapy often becomes ineffective due to the emergence of drug resistance, which remains a major obstacle in achieving long-term treatment success. Tumor cells can adopt multiple adaptive strategies that allow them to withstand the action of anticancer agents. These strategies include the overexpression of drug efflux pumps such as ATP-binding cassette (ABC) transporters, mutations that modify drug-binding sites, increased ability to repair damaged DNA, and the activation of alternate survival pathways. Epigenetic changes—including variations in DNA methylation patterns, histone remodeling, and regulation by microRNAs—further support resistant phenotypes. Additionally, the tumor microenvironment contributes significantly by creating conditions such as hypoxia, altered metabolic states, and interactions with stromal and immune cells that collectively reduce drug sensitivity. Cancer stem cells (CSCs), known for their high survival capacity, also play a crucial role in promoting multidrug resistance and tumor regrowth. A comprehensive understanding of these mechanisms is vital for developing improved therapeutic approaches and overcoming resistance in cancer treatment. This review discusses the major biological processes responsible for drug resistance and highlights potential strategies to enhance therapeutic effectiveness.
Cancer therapy has progressed remarkably over the past decades, yet the development of drug resistance continues to limit the long-term success of treatment. Many patients initially respond well to chemotherapy, targeted therapy, hormonal therapy, or immunotherapy, but tumors often adapt and become unresponsive, leading to disease recurrence and treatment failure (1,2). Drug resistance may be present from the beginning (intrinsic resistance) or may arise after repeated exposure to anticancer agents (acquired resistance). This ability of cancer cells to survive therapeutic pressure reflects a wide range of molecular, genetic, and environmental adaptations (3). A key contributor to resistance is the increased activity of efflux transporters, particularly ATP-binding cassette (ABC) proteins, which actively pump anticancer drugs out of tumor cells and reduce their intracellular concentration (4). Mutations or structural changes in drug targets, such as receptor tyrosine kinases, also reduce the effectiveness of targeted therapies (5). Epigenetic alterations—including DNA methylation changes, chromatin remodeling, and microRNA regulation—further influence cellular behavior and contribute to therapy evasion (6). In addition to these intrinsic cellular mechanisms, external factors within the tumor microenvironment, such as hypoxia, stromal interactions, and altered metabolic conditions, create protective niches that diminish the sensitivity of cancer cells to treatment (7). The presence of cancer stem cells (CSCs), a small population of highly resilient cells capable of self-renewal, also plays a central role in multidrug resistance and tumor regrowth (8).
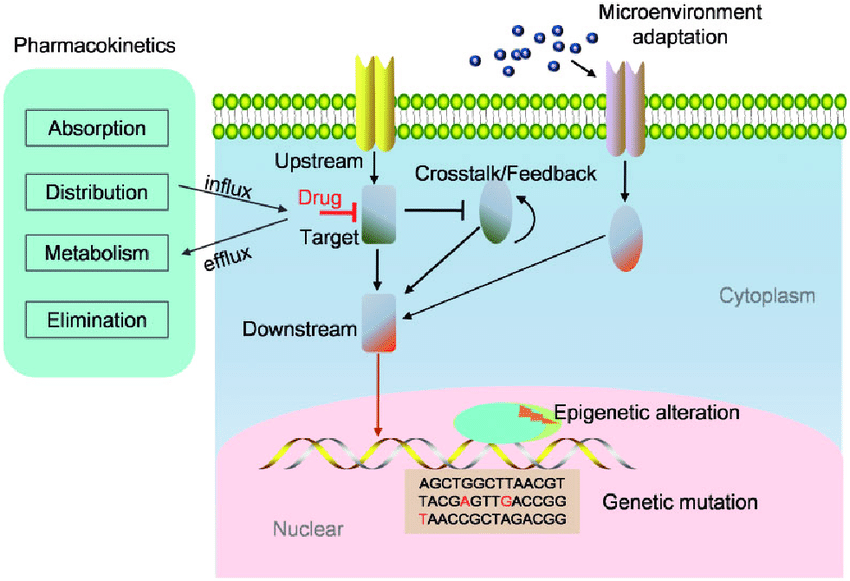
Fig 1
Pathophysiology:
The pathophysiology of drug resistance in cancer reflects a complex set of adaptive responses that help tumor cells survive therapeutic stress. One of the most prominent mechanisms involves the increased activity of ATP-binding cassette (ABC) transporters, such as P-glycoprotein, which actively expel anticancer drugs from the intracellular space, lowering drug levels to sub-therapeutic concentrations (9,10). Resistance also arises from mutations within critical drug-interacting proteins, including receptor tyrosine kinases, topoisomerases, and cytoskeletal components, resulting in diminished drug binding and loss of treatment efficacy (11). Tumor cells may additionally upregulate DNA repair pathways such as homologous recombination and nucleotide excision repair, allowing them to quickly correct therapy-induced DNA damage and tolerate DNA-targeting agents (12).
Epigenetic changes further contribute to resistance. Alterations in DNA methylation, histone structure, and microRNA expression can shift gene regulation in favor of cell survival, reduced apoptosis, and adaptation to ongoing drug exposure (13). Many cancer cells enhance survival by activating compensatory signaling networks—such as PI3K/Akt, MAPK/ERK, and NF-κB—that maintain proliferation even when primary pathways are inhibited by treatment (14). The tumor microenvironment (TME) also plays an essential role; conditions such as low oxygen levels, acidic pH, cytokine release, and supportive stromal interactions create a physical and biochemical environment that protects tumor cells and decreases drug responsiveness (15).
A distinct population known as cancer stem cells (CSCs) adds another layer to the resistance process. CSCs possess strong DNA repair capacity, low proliferative rates, and high survival potential, making them significantly more tolerant to chemotherapy and targeted therapy. Their persistence promotes tumor regrowth and is closely associated with multidrug-resistant behavior (16).
Role of Various Cellular Pathways and Receptors in Drug Resistance:
Mechanisms of Drug Resistance in Cancer Therapy: Roles of Cellular Pathways and Receptors
Alterations in EGFR, HER2, MET, and IGF-1R—such as mutations or amplification—enable tumor cells to bypass targeted therapies. EGFR T790M/C797S mutations and HER2 overexpression reactivate downstream signaling and lead to resistance to TKIs and monoclonal antibodies (17,18).
This survival pathway becomes hyperactivated through PI3K mutations or PTEN loss. Increased AKT signaling enhances anti-apoptotic proteins and metabolic adaptation, contributing to resistance to chemotherapy, radiotherapy, and targeted agents [17,19].
Secondary mutations in RAS or BRAF allow reactivation of MAPK signaling despite inhibitor therapy. Cross-talk with the PI3K pathway enables rapid compensatory survival, causing resistance to EGFR and BRAF inhibitors (18,19).
Overexpression of ABCB1 (P-gp), ABCC1, and ABCG2 pumps drugs out of cells, lowering intracellular concentration and producing multidrug resistance. This mechanism affects many chemotherapies and TKIs (20).
Loss of TP53 function or upregulation of BCL-2 family proteins reduces apoptosis, enabling cancer cells to survive treatment. Enhanced DNA repair mechanisms (HR, NHEJ) also reduce the effectiveness of DNA-damaging agents and PARP inhibitors (17,21).
EMT and CSC pathways (Wnt, Notch, Hedgehog) increase plasticity, drug efflux, and survival. Hypoxia (HIF-1α) and cytokines in the tumor microenvironment promote resistance by activating STAT3 and TGF-β pathways, creating protective niches (21,22).
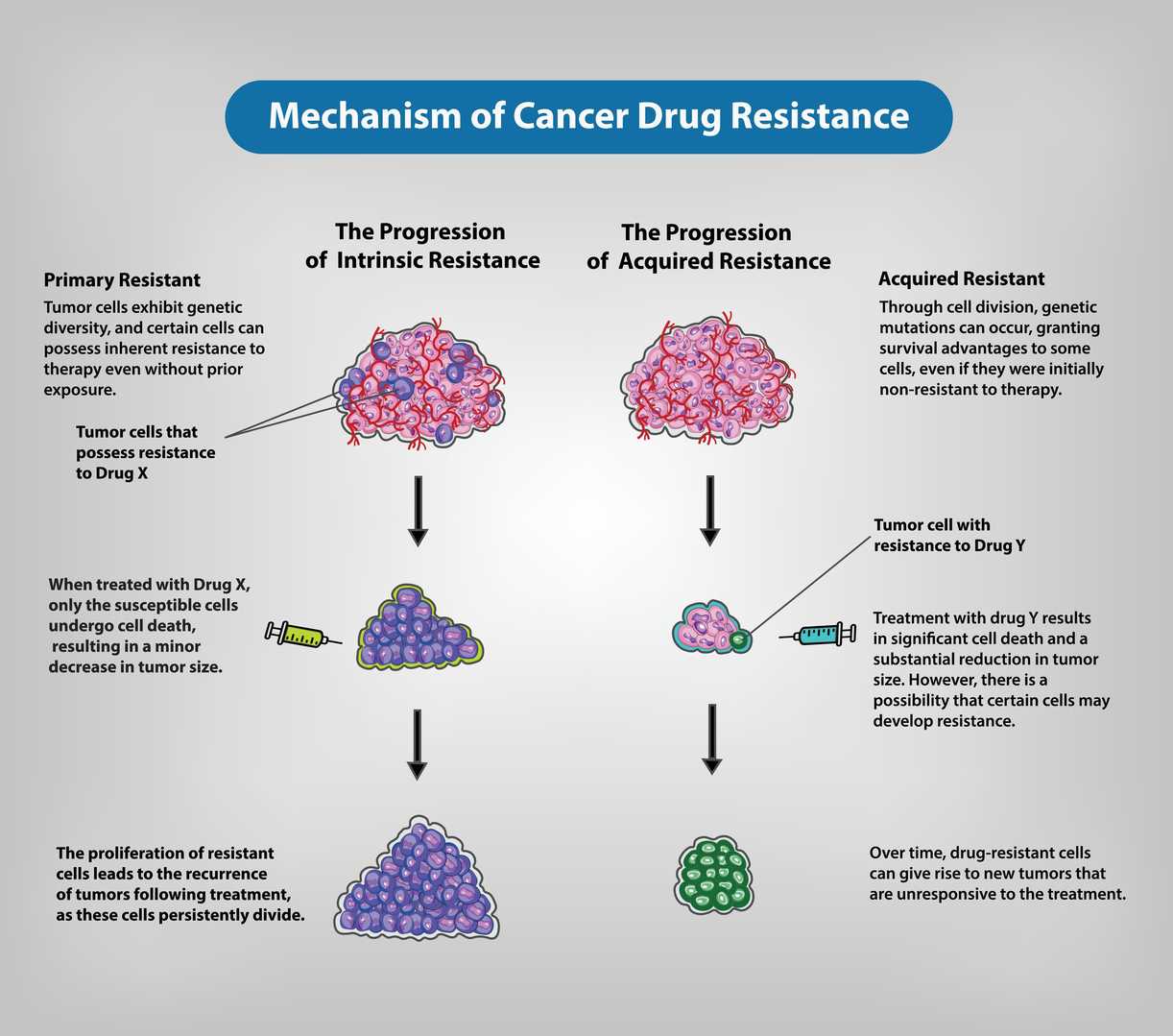
Fig 2
New Therapeutic Strategies for Overcoming Drug Resistance:
Using more than one targeted agent at the same time can suppress the feedback loops that tumors use to survive treatment. Approaches such as co-inhibiting the MAPK and PI3K/AKT pathways, or pairing EGFR inhibitors with MET or HER2 blockers, can delay or reverse resistance (23,24).
Nanoparticle formulations help deliver drugs deeper into tumor tissue and reduce recognition by ABC transporters, which normally pump drugs out of cancer cells. This strategy improves intracellular drug retention and reduces multidrug resistance (25).
That inhibit anti-apoptotic proteins (e.g., BCL-2) or reactivate p53 signaling can restore programmed cell death in resistant tumors. BH3 mimetics are particularly effective in sensitizing cancer cells to treatment (26).
Blocking multiple DNA repair pathways at once—such as combining PARP inhibitors with ATR or CHK1 inhibitors—prevents cancer cells from compensating for DNA damage and improves response in PARP-resistant cancers (27).
Drugs that interfere with Wnt, Notch, Hedgehog, or TGF-β signaling can reduce tumor plasticity and stem-like properties, making resistant cancer cells more vulnerable to therapy (28).
Pairing immune checkpoint inhibitors (e.g., anti-PD-1, anti-PD-L1, anti-CTLA-4) with conventional treatments such as chemotherapy or radiation enhances anti-tumor immunity and helps overcome (29).
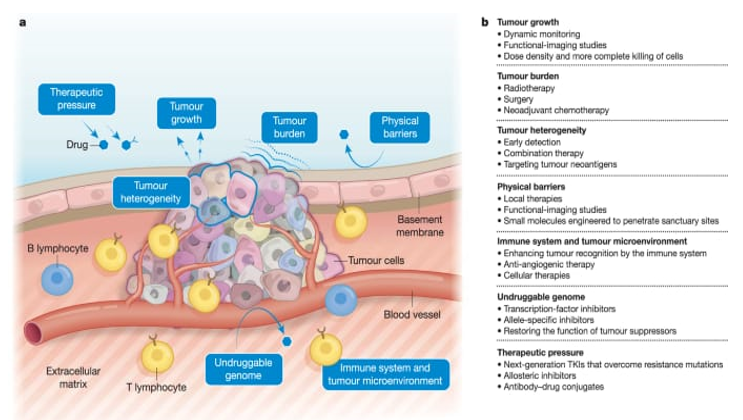
Fig 3
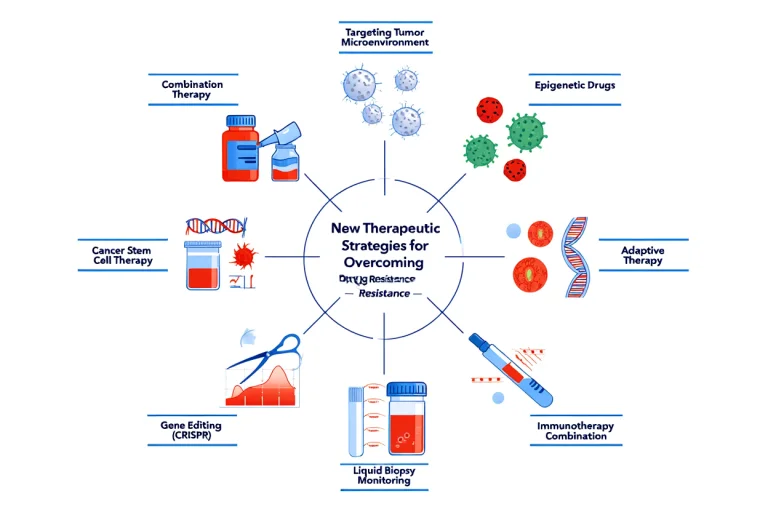
Fig 4
ACKNOWLEDGEMENTS:
The authors would like to express their sincere appreciation to all researchers whose valuable work has contributed to the understanding of drug-resistance mechanisms in cancer therapy. We also extend our gratitude to our academic mentors and colleagues for their constructive guidance and continuous encouragement throughout the development of this review. Their insights greatly enhanced the clarity and depth of this manuscript. Finally, we acknowledge the support of our institution for providing the resources necessary to complete this work.
CONCLUSION:
Drug resistance in cancer results from a combination of genetic changes, pathway reactivation, altered drug transport, and support from the tumor microenvironment. These interconnected mechanisms allow cancer cells to survive and limit the effectiveness of therapy. Addressing resistance will require combining different treatment strategies and using molecular tools to guide personalized care. Continued research into these mechanisms is essential for developing therapies that achieve longer and more durable responses.
REFERENCES



Komal Dhatrak*, Diksha Lokhande, Prajwal Gosavi, Mechanisms of Drug Resistance in Cancer Therapy, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 239-245 https://doi.org/10.5281/zenodo.17785778
 10.5281/zenodo.17785778
10.5281/zenodo.17785778
