Advance Institute of Biotech & Paramedical Sciences, Kanpur.
The quantitative identification of medications and their metabolites in biological fluids is known as bioanalysis. This method is applied very early in the medication development phase to support programs that investigate the pharmacokinetics and metabolic metabolism of chemicals in living cells and animals. A number of excellent review articles addressing different scientific and technological facets of bioanalysis have been added to the literature in recent years. Nowadays, it is generally acknowledged that bioanalysis plays a crucial role in the pharmacokinetic and pharmacodynamic characterisation of a new chemical entity from the moment of discovery through the many phases of drug development that culminate in its approval for sale. The function of bioanalysis in the creation of pharmaceutical drugs is examined, with an emphasis on the specific tasks carried out at each phase of the process and the range of sample preparation matrices that are encountered. Examples of how these high throughput requirements are being satisfied in bioanalysis are shown, along with recent advancements and industry trends for quick sample throughput and data creation.
A new medicine's development and discovery can cost up to $1 billion, and it may take ten years for the drug to be available for purchase.[1] The process of creating compounds and assessing each one's characteristics in order to ascertain whether only one new chemical entity (NCE) may be chosen to become a safe and effective medication is known as drug discovery and development [2]. It has also been proven that toxicokinetics is a crucial component of toxicity testing [3]. A sensitive and targeted bioanalytical method is crucial given the emphasis on PK/toxicokinetics and the higher potencies of the newer medications [4]. It is commonly known and acknowledged throughout the world that bioanalysis has emerged as a crucial tool in the drug discovery and development process [5]. Additionally, there are numerous analytical techniques accessible for both generic and prescription medications [6-7].A useful guidance for early clinical programs is the bioanalytical data produced in discovery and preclinical studies [8–9]. These comparisons are especially helpful in the initial dose escalation phase one investigation. In order to optimize this, we routinely generate PK data in between dose increases [10].
Bioanalysis:
The quantitative assessment of a substance or its metabolite in biological fluids, mainly blood, plasma, serum, urine, or tissue extract, is commonly referred to as bioanalysis [11]. Drug concentration in the biological matrix should be accurately measured by bioanalytical techniques that are sensitive, specific, repeatable, and accurate [12]. The complicated biological sample composition, low drug concentration, small sample volume, high variability among some patients at different timepoints, and having different subjects in the same group are some of the challenges related to bioanalytical assays [13]. Usually, chromatographic assays and ligand binding assays are used to detect and quantify the medicines in biological samples [14].
3.1-Chromatographic Assay:
Although a variety of detectors, such as ultraviolet, fluorescence, and chargrd aerosol detection, can be coupled with high performance liquid chromatography (HPLC) and ultra high performance liquid chromatography (UPLC), the gold standard is currently thought to be HPLC & UPLC coupled with liquid chromatography and mass spectrometric detection (LC-MS/MS). The standard analytical technique for measuring medications, peptides, and oligonucliotides in biological materials is LC-MS/MS [13]. Protein precipitation, solid phase extraction, and liquid-liquid extraction are frequently employed pretreatment techniques. During sample analysis, sample cleanup might also be the most labor-intensive and time-consuming phase. Therefore, would be a crucial factor to take into account while choosing a sample cleansing technique. [15]
This approach does, however, have several disadvantages. For precise detection and quantification of every analyte, the LC-MS/MS method employs an internal standard. An isotope-labeled version of the target analyte, known as a stable isotopes-labelled internal standard (SILIS), is the ideal internal standard [16].
Unfortunately, it can take a lot of time and money to synthesize the SILIS. Isobaric substances that co-elute at the same mass-to-charge ratio (m/z, the unit in which a mass spectrometer detects analytes) and fragment exactly like the analyte in the MS/MS may also be present in the sample. If so, these contaminants need to be separated chromatographically before being detected by MS/MS. Examples of such separations are size-exclusion chromatography or reverse phase separation combined with ion exchange chromatography [17].
3.2-Ligand Binding Assay:
An LBA assesses the binding interaction between the analyte and the binding target indirectly rather than directly. One of the most used plateforms for LBAs is the enzyme linked immunosorbent assay (ELISA). The four primary ELISA types vary in how the antigen or antibody is deposited onto the plate and how the signal is picked up [18–19].
3.2.1- Direct ELISA: A primary antibody that is specific to the antigen is incubated with the antigen after it has been directly adhered to the microtiter plate's wells. An enzyme for detection is pre-labeled onto the main antibody.
3.2.2- Indirect ELISA: A primary antibody that is specific to the antigen is incubated with the antigen after it has been adhered to the microtiter plate's wells. For detection, a secondary antibody that has been labeled against the primary antibody's host species attaches itself to the primary antibody.
3.2.3- Sandwich ELISA: Two antibodies that are specific to distinct antigen epitopes are needed for this format. The plate surface is coated with one of the antibodies, which then binds to the antigen. The antigen can be detected more easily thanks to the other antibody.
3.2.4- Competitive ELISA: This format involves an unlabeled sample antigen or antibody and a reference antigen or antibody competing for the same binding site. This format is compatible with all of the previous ELISA formats.The reference and sample antigens compete with one another to attach to the labeled antibody.
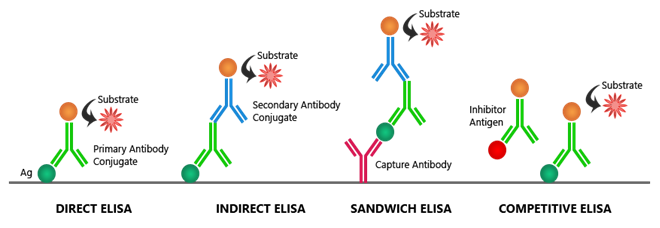
Fig:1- Types of ELISA Assay
The concentration of antigen in the sample is inferred using a calibration curve with a known concentration of reference antigen. Both colorimetric and fluorimetric detection signals are possible for each type of ELISA [18–19].
Validation Parametrs:
4.1-Specificity/Selectively -The terms "specificity" and "selectively" are used interchangeably in bioanalytical validation. The ability of the bioanalytical technology to yield a single analyte is referred to as specificity. On the other hand, selectivity refers to a method's capacity to distinguish an analyte of interest from another analyte [20].
4.2- Accuracy: An analytical method's accuracy indicates the actual value or concentration of an analyte.
4.3- Precision: When the process is repeated on several aliquots of a single homogenous volume, precision is the proximity of individual analyte measurements [21–22].
Quantiation limit:The quantification limit is the lowest concentration of analyte in the sample as determined by certain analytical techniques.
4.4- Detection Limit: The smallest quantity of an analyte that can be detected but not measured is known as the detection limit [23].
4.5- Ruggedness: The degree of reproducibility of the test result achieved following sample analysis under a range of typical test settings is known as the "ruggedness" of an analytical or bioanalytical process [24].
4.6- Stability: This refers to an analyte's chemical stability under particular circumstances. The purpose of the stability test is to identify any analyte degradation that may occur during the sample collection, processing, storage, preparation, and analysis phases [25].
Drug Discovery/ Design:
The goal of bioanalysis during the drug discovery phase is to supply acceptable concentrations of compounds that will be utilized for lead component identification and discrimination. As a result, the analyst's goal at this point should be to provide a quick, easy approach for identification with a high throughput [26]. Finding and characterizing new targets as well as synthesizing and screening novel lead molecules are all part of the drug discovery process [27]. Once the compound has the necessary biological activity and is appropriate for drug development, several analogs or chemically similar compounds will be produced and tested. Physical-chemical characteristics including solubility, lipophilia, and stability are assessed in the subsequent screening phase. Predicting protein binding, tissue distribution, and absorption in the gastrointestinal tract depends on these measurements [28]. ADME characteristics are the collective term for these tests. Information regarding overall drug deposition and progress is provided by these research [29].
Drug Development:
The process of introducing a new pharmaceutical medication to the market when a lead ingredient is found is known as drug development. Preclinical studies on microbes and animals are included. For an investigational novel medicine to start human clinical trials, it is crucial that the application for regulatory status be filed. The assessment of novel drug molecules' safety, toxicity, and effectiveness as well as the medication's pharmacokinetic characteristics are the main goals of drug development [30].
6.1-Preclinical Phase: Drug discovery produces molecules that are lead candidates for novel chemical entities. They show promise in combating a specific biological target, which is crucial in the fight against a given illness. First, the lead component's safety, toxicology, pharmacokinetics, and metabolism in humans are examined. The physical characteristics of the lead component, such as its chemical composition, stability, and solubility, must be taken into account throughout drug development. To meet the regulatory requirements of drug licensing authorities, drug development must be completed satisfactorily. The data gathered during preclinical testing was submitted as a new drug application to the drug regulatory body. Development proceeds to the clinical stage following this approval [31–32].
6.2- Clinical trials: It involves mainly 4 phases.
6.2.1- Phase-I: Healthy volunteers are used in Phase I to determine safety and dosage.
6.2.2-Phase-II: Small numbers of patients with a specific condition and an initial readout of a new medication entity are used in phase-II.
6.2.3-Phase-III: Although it is much larger, phase III is comparable to phases I and II. At this point, it is crucial to assess the safety and effectiveness of the treatment on a significant number of patients. Testing may end at this point and the lead component may be prepared for the new drug application stage if safety and efficacy are established by the regulatory agency.
6.2.4- Phase-IV: A post-marketing monitoring study known as phase-IV is conducted under restrictions set by the Food and Drug Administration.
During medication development, the majority of novel drugs fail because they either have unacceptable toxicity or do not exhibit the desired effect in clinical trials against the targeted disease [33–34].
Abbreviations:
ADME- Absorption, Distribution, Metabolism, Excretion. ELISA- Enzyme Linked Immuno Sorbent Assay. FDA- Food and Drug Administration. HPLC- High Performance Liquid Chromatography. SILIS- Stable Isotopes Labeled Internal Standard. UPLC- Ultra-High-Performance Liquid Chromatography. PD- Pharmacodynamics. PK- Pharmacokinetics. LBA- Ligand Binding Assay.
CONCLUSION:
Bioanalytical techniques are essential for drug research and discovery as well as for evaluating PK, PD, and toxicity data. This helps with regulatory decision-making and facilitates the assessment of a drug's safety and effectiveness. Before the validation research begins, a validation plan should explicitly define the acceptance criteria. The developed assay should be robust enough to allow for minor modifications and easy adoption to meet other bioanalytical needs, such as characterization of the metabolites' plasma levels, acceptability to a toxicokinetic study, and acceptability to a drug-drug interaction study. It was discussed what regulations apply to the development and validation of bioanalytical methods.
REFERENCES


Vivek Vishnoi*, Dr. Abhinav Mishra, Ashish Kumar, Namit Kumar, Shreya Chaurasiya, Importance of Bioanalysis in Drug Discovery & Development: A Comprehensive Review, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 01, 62-68. https://doi.org/10.5281/zenodo.14584659
 10.5281/zenodo.14584659
10.5281/zenodo.14584659
