YSPM`s Yashoda Technical Campus, Faculty of Pharmacy, Wadhe, Satara, Maharashtra, India.
Harmonizing regulatory submissions is vital for efficient global drug development and approvals. The Common Technical Document (CTD) and Electronic Common Technical Document (eCTD) created by the International Council for Harmonization (ICH) provide a unified global standard. This standard allows for a consistent dossier structure, smoother regulatory reviews, and simultaneous submissions in different regions. Over the past two decades, the CTD has changed from a paper-based system to a highly structured digital submission model (eCTD v3.2.2), and it is now evolving into an interoperable messaging-based standard (eCTD v4.0). Major regulatory agencies, including the USFDA, EMA, PMDA, Health Canada, TGA and Swiss medic have mandated eCTD for most application types demonstrating a global commitment to harmonization.As drug development becomes more complex, Artificial Intelligence (AI) tools have emerged as transformative technologies. These tools can automate document structuring, intelligently tag metadata, validate content, perform quality reviews, hyperlink automatically, and manage lifecycles predictively. AI systems minimize human error, streamline document compilation, and speed up approval timelines.
Global harmonization in pharmaceutical regulation involves aligning technical, quality, safety, and efficacy standards across countries. This ensures that medicinal products meet uniform requirements worldwide. Harmonization leads to faster approvals, reduces duplication, and improves patient access to high-quality medicines. The founding of the International Council for Harmonization (ICH) in 1990 was a significant step toward unifying regulatory expectations 1.
Before harmonization, companies had to create separate dossiers for each regulatory region, often facing major differences in format and data requirements. Studies show this caused inefficiencies, increased regulatory burdens, and delays in patient access 2.
The ICH introduced a series of structured guidelines categorized as Quality (Q), Safety (S), Efficacy (E), and Multidisciplinary (M). Among these, the ICH M4 guideline standardized the CTD, while the ICH M8 guideline introduced the eCTD, establishing the universal foundation for global submissions 3-4.
Harmonization enhances consistency and credibility in regulatory decision-making worldwide. Studies indicate that using harmonized CTD/eCTD formats boosts efficiency, regulatory communication, and transparency in scientific reviews 1-2.
Before the CTD was introduced, pharmaceutical companies had to prepare multiple dossier formats for Europe, the United States, and Japan. Each region required distinct administrative structures, scientific data formats, and presentation layouts. This led to duplication, higher costs, longer timelines, and inconsistent scientific evaluations. To solve this fragmentation, the ICH created the CTD in 2000. This initiative transformed drug submissions by establishing a universal dossier format that eliminated formatting differences and allowed simultaneous submissions across multiple markets. The next step was the eCTD introduced in 2003. This allowed electronic transmission, digital navigation, real-time lifecycle management, XML backbone structure, and automated validation. Today, eCTD is mandatory in most heavily regulated markets, and its global usage continues to grow.
As regulatory requirements change, traditional manual compilation methods are no longer sufficient. The vast amounts of quality, nonclinical, and clinical data require smart tools for structuring, formatting, linking, and validating documents. As a result, AI-based systems have become essential for ensuring accuracy, speed, and global compliance in eCTD submissions.
ICH (INTERNATIONAL COUNCIL FOR HARMONIZATION)
Establishment & Objectives:
The ICH was established in 1990 by regulatory authorities and pharmaceutical industries from the US, Europe, and Japan. Its goal is to standardize technical requirements for drug registration globally. The main objectives include reducing duplication of clinical and nonclinical data, harmonizing guidelines, and improving global drug availability 3.
ICH Guidelines Related To CTD & eCTD:
The key guidelines governing CTD/eCTD formats include:
M4 – ICH M4 defines the structure, format, and content of the Common Technical Document (CTD). Purpose of ICH M4: Create a single, harmonized format for drug registration dossiers; Support simultaneous submissions in multiple markets; Improve communication between regulatory authorities and applicants 3.
M2 – ICH M2 provides technical standards and electronic specifications for transferring regulatory information. It ensures that documents submitted electronically are compatible, interoperable, and readable across different regulatory agencies.
Purpose of ICH M2: Develop global technical standards for electronic submissions; Enable agencies to receive CTD data digitally; Prepare the foundation for eCTD and future digital submissions 3.
M8 – ICH M8 sets the global electronic submission standard for eCTD. It details the technical requirements for compiling, organizing, submitting, and maintaining CTD data electronically. These guidelines ensure uniformity in dossier structure, electronic transmission, metadata requirements, file formats, and lifecycle management.
Purpose of ICH M8: Convert the paper CTD format (M4) into a fully digital format; Harmonize electronic submissions across regions; Support hyperlinking, bookmarks, metadata, and XML structure; Improve submission quality, speed, and transparency 3.
Role In Global Harmonization:
The ICH guidelines allow pharmaceutical companies to create a single standardized dossier for multiple regulatory agencies. This reduces time and costs for global submissions. Harmonization strengthens regulatory collaboration, promotes consistent scientific decision-making, and supports efficient drug approvals worldwide.
EVOLUTION FROM CTD TO ECTD
The shift from CTD to eCTD happened over several phases:
Year 2000: Introduction Of CTD
A paper-based CTD with five standardized modules was released to simplify dossier structure across regions.
Year 2003 – 2005: Transition To eCTD V3.0
The introduction of electronic submissions enabled hyperlinking, bookmarks, validation, and lifecycle management.
Year 2008 And Onwards: Adoption Of eCTD V3.2.2
This version became widely accepted and mandatory in major regions, enhancing compatibility and technical specifications.
Current Phase: eCTD V4.0 (RPS-Based)
The latest version uses Health Level Seven (HL7) standards. It enables message-based communication, better interoperability, and advanced metadata features.
Table No. 1: Evolution Of CTD To eCTD
|
Version |
Year |
Key Features |
|
CTD |
2000 |
Paper format, 5 modules |
|
eCTD v2.0 |
2003 |
Basic electronic transitions |
|
eCTD v3.0 |
2005 |
XML backbone introduced |
|
eCTD v3.2.2 |
2008 |
Global standard, lifecycle rules |
|
eCTD v4.0 |
2021 |
HL7 RPS, messaging-based |
STRUCTURE AND COMPONENTS OF CTD AND ECTD
The CTD structure organizes regulatory information into five standardized modules, which allows for a harmonized review across global authorities. Research confirms this system improves document organization, review efficiency, and consistency across regions 1-2.
ICH M4 standardizes the CTD into five modules, which are:
1. Module 1 – Regional Administrative Information (not harmonized)
2. Module 2 – Summaries and Overviews
3. Module 3 – Quality (CMC)
4. Module 4 – Nonclinical Reports
5. Module 5 – Clinical Study Reports
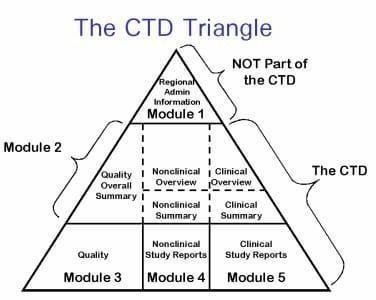
Figure No. 1: CTD Triangle
CTD Structure (5 Modules):
Module 1 – Regional Administrative Information:
Module 1 contains administrative and product-specific information required by each regulatory authority, which differs among regions. Agencies like the FDA, EMA, PMDA, CDSCO, and Health Canada publish specific Module 1 requirements 5-6.
Module 2 – Summaries And Overviews:
Module 2 offers high-level summaries of Modules 3, 4, and 5. It includes the Quality Overall Summary (QOS), Nonclinical Overview, Clinical Overview, and both tabulated and written summaries. This module plays a crucial role as it connects raw data to regulatory assessment, aiding in benefit-risk evaluations 1-2.
Module 3 – Quality (CMC):
Module 3 contains chemistry, manufacturing, and controls (CMC) information. It represents one of the most scientifically intensive sections of the CTD. It includes: API characterization, Drug product development, Manufacturing process, Specifications, Validation, and Stability studies. Research shows that inconsistencies in Module 3 are common causes of regulatory queries worldwide 1-2.
Module 4 – Nonclinical Studies:
Module 4 covers toxicology and pharmacology studies done in animals. It includes: Acute, subacute, and chronic toxicity; Genotoxicity; Reproductive toxicity; Carcinogenicity; and Pharmacokinetic and pharmacodynamic studies. A standardized Module 4 structure improves the comparability of toxicology findings across regions 2.
Module 5 – Clinical Study Reports:
Module 5 includes all human clinical trial data from Phase I to III studies, bioequivalence studies, observational research, and post-marketing data. Research indicates that Module 5 is the most significant for assessing drug efficacy and safety. Its strict formatting enhances review team efficiency 1.
eCTD Technical Features:
XML Backbone And Metadata Framework:
The XML backbone is the core technical component of the eCTD. This automatically generated file functions as the "central nervous system" of the submission. It contains detailed metadata about each document, such as its location, document type, lifecycle operation, title, and heading level. Reviewers rely on the XML backbone for automated navigation, indexing, and validation, rather than manually navigating PDFs. The XML file ensures that the eCTD follows the structure outlined by ICH M8. It also encodes relationships between documents, such as which files replace older versions or supplement existing information. This metadata-driven architecture makes the eCTD machine-readable. As a result, regulatory authorities can use validation tools and automated review systems. The XML backbone ensures that regulatory submissions are consistent, minimize errors, and are easy to interpret, no matter what software or region is used to view them.
Lifecycle Management Using Sequences:
A key feature of eCTD is its strong lifecycle management system. Unlike paper submissions that need physical replacement or manual version control, the eCTD organizes the entire product lifecycle into sequences. A sequence is a set of documents submitted to the agency for a specific regulatory activity. Each document in a sequence has a lifecycle operator that defines how it should be interpreted. The reviewer's software merges all sequences to create the Current View, displaying only the most recent version of every file. This cuts down confusion, keeps versioning accurate, and simplifies inspection and auditing. Lifecycle management is a significant advantage of eCTD. It allows regulators to track product information from development through to post-marketing updates.
Structured Folder Architecture:
Even though eCTD is based on the CTD modules, it has strict rules on how documents should be organized in folders. The hierarchical structure is standardized globally, promoting uniformity and predictability. This arrangement ensures that submissions meet international expectations, making it easier for regulators to find information without digging through inconsistently organized material. The structured design also improves transparency and makes automated validation easier.
PDF Publishing Standards (File Properties, Bookmarks, And Hyperlinks):
The eCTD requires documents to be published as high-quality, compliance-ready PDF files that adhere to strict standards. These PDF documents must have internal bookmarks for every major heading and subheading. These bookmarks help reviewers quickly navigate lengthy reports, such as clinical or nonclinical studies. The requirement for hyperlinks is just as important. Hyperlinking turns submissions into a well-organized document ecosystem, which significantly reduces review time.
Validation Requirements And Technical Conformance:
Every eCTD submission must undergo technical validation before acceptance. Agencies like the FDA, EMA, PMDA, and Health Canada use automated tools to check compliance with ICH M8 and regional specifications. If errors are found, the submission is either rejected or put on hold until corrected. This process ensures that only high-quality, consistent, machine-readable submissions are accepted, avoiding delays during the regulatory review. Technical validation is therefore essential for maintaining standardization.
Electronic Gateway Transmission:
eCTD submissions are sent electronically to regulatory authorities through secure gateways: FDA ESG (Electronic Submissions Gateway), EMA CESP (Common European Submission Portal), Health Canada CESG, PMDA eGateway, TGA Submission portal, and India – SUGAM or future eCTD portal. Gateway transmission is a crucial technical aspect that makes regulatory communication a fully digital and traceable process.
Reviewer-Friendly Electronic Navigation:
Regulatory reviewers use specific eCTD viewing software (like Lorenz DocuBridge or Extedo eCTD viewer) that displays the dossier as a collapsible tree structure. This electronic navigation tree greatly improves efficiency, especially for large NDAs or MAAs that contain thousands of documents.
Global Standardization And Interoperability:
eCTD is standardized worldwide across ICH regions. Thanks to standardized metadata and XML schema, eCTD software can be used consistently in different regions, supporting global drug development strategies.
Transition To eCTD V4.0 (HL7 RPS Standard):
eCTD v4.0 brings new technical features. It represents a shift toward a more structured, machine-readable regulatory environment. This change enables integration with AI, automation, and real-time data validation.
GLOBAL IMPLEMENTATION OF CTD AND ECTD
Global regulatory agencies have adopted CTD and eCTD at different rates, but most major markets now require their use. Comparative research from the USA, EU, Japan, and China shows the widespread acceptance of CTD/eCTD as the universal submission standard 7.
United States (FDA): United States Food And Drug Administration
The U.S. FDA requires eCTD for NDA, ANDA, BLA, DMF, and IND submissions. The FDA's Electronic Submissions Gateway (ESG) and eCTD Technical Conformance Guide outline the detailed structure, metadata, and validation requirements 5. The FDA is one of the strictest agencies about technical compliance, automatically rejecting invalid submissions due to hyperlink or XML errors 8.
European Union (EMA): European Medicines Agency
The EMA requires eCTD for centralized procedures and uses the CESP portal for transmission. The EMA is also an early adopter of structured data standards and a strong supporter of eCTD v4.0 adoption 9. The EMA's harmonized approach improves regulatory efficiency among member states, as shown in several reviews 2.
Japan (PMDA): Pharmaceuticals And Medical Devices Agency
Japan's PMDA has long supported CTD and eCTD. It rigorously enforces metadata and XML validation rules, maintaining high technical standards. Comparative studies indicate that PMDA's eCTD implementation is among the most developed worldwide 7.
India (CDSCO): Central Drugs Standard Control Organisation
India uses the CTD format under NDCTR 2019 and is moving toward mandatory eCTD. Regulatory policy papers indicate that CDSCO is transitioning to full digital submission processes, in line with global harmonization efforts 6.
Canada (Health Canada): Canada's Federal Health Regulatory Authority
Health Canada mandates eCTD for major submissions using the CESG portal and closely matches FDA & EMA eCTD specifications 10.
Table 2. Regulatory Body Requirement For eCTD
|
Region |
Regulatory Body |
Status |
|
USA |
FDA |
Mandatory |
|
EU |
EMA & NCA |
Mandatory |
|
JAPAN |
PMDA |
Mandatory |
|
Canada |
HC |
Mandatory |
|
Australia |
TGA |
Accepts eCTD |
|
India |
CDSCO |
Transitioning to eCTD |
CHALLENGES & LIMITATIONS IN CTD AND ECTD PREPARATION
Data Fragmentation Across Teams:
In pharmaceutical companies, preparing a CTD/eCTD dossier requires coordinated input from multiple departments, including R&D, Formulation, Analytical Development, QA, QC, Manufacturing, Clinical, Non-clinical, and Regulatory Affairs. Each team generates vast amounts of scientific data independently, often in different formats with varying documentation standards. This leads to data fragmentation, as important information becomes scattered across multiple systems, personal drives, emails, and paper records. Such fragmentation complicates finding the latest document versions, leading to mismatches between data sets, incomplete modules, and inconsistencies in the dossier.
Technical Errors In eCTD:
eCTD is highly technical and requires adherence to strict guidelines concerning structure, detail, metadata, and document formatting. Technical errors can occur when publishers do not follow the ICH M8 specifications precisely. Common errors include incorrect XML backbone creation, invalid or missing metadata, broken hyperlinks, PDFs without proper bookmarks, incorrect leaf titles, or outdated PDF versions. Issues with PDF optimization, like non-searchable text, poorly scanned images, or oversized files, also result in rejections. Technical mistakes are not just superficial; they hinder reviewers' ability to navigate the dossier smoothly. For instance, a broken hyperlink or incorrect lifecycle operator (new/replace/delete) can disrupt the review process and halt technical validation. Some regulatory authorities automatically reject submissions when technical validation fails, necessitating a full republication of the sequence. Because eCTD is lifecycle-based, even small mistakes can disrupt links across sequences. Preparing eCTD requires specialized software, trained publishers, and multi-level validation before final submission.
High Cost Of eCTD Publishing Systems:
One major obstacle for many pharmaceutical firms, particularly generic manufacturers and smaller companies, is the expensive nature of eCTD publishing systems. Commercial publishing tools, like Lorenz DocuBridge, EXTEDO, and Freyr SUBMIT, come with significant licensing fees, annual maintenance costs, and the need for advanced hardware. In addition, companies must invest in secure servers, document management systems (DMS), XML publishing tools, and PDF processing software. Beyond software expenses, organizations face costs for staff training, IT support, and periodic software updates to comply with new regulatory standards. For smaller firms, these expenses lead to considerable financial strain. Many struggle to maintain dedicated publishing teams or acquire multiple licenses for various departments. Outsourcing eCTD publishing to regulatory consultants can lighten the internal load, but it increases financial reliance on external partners. Therefore, the high costs associated with publishing tools and infrastructure remain a significant hurdle for adopting eCTD systems, especially in resource-limited situations.
Frequent Regulatory Updates:
Regulatory agencies frequently update their submission standards, templates, and technical specifications. With shifts in regional requirements, updated validation criteria, and constant Q&A updates, companies face the ongoing challenge of staying current with these changes. Each update requires adjustments to internal SOPs, templates, publishing settings, and validation procedures. Failing to adapt promptly often results in non-compliant submissions, technical rejections, or additional deficiency letters. Furthermore, different regions implement changes at varying times, complicating efforts to maintain global regulatory coherence. A dossier that meets European standards may require structural changes to meet FDA or PMDA requirements. Regulatory intelligence teams must continuously track updates, participate in training sessions, and accurately interpret new requirements. This ongoing need for regulatory adaptation adds to the workload, drives system modifications, and increases complexity in regulatory affairs departments.
Human Resource Limitations:
Preparing CTD/eCTD documents is a specialized task that requires skills in regulatory science, document management, scientific writing, lifecycle management, and technical publishing. However, many pharmaceutical companies struggle with shortages of trained regulatory professionals. Regulatory affairs teams tend to be small, with limited staff managing multiple major projects at once. This results in heavy workload pressure, longer hours, and a greater chance of errors in documentation. Inexperienced staff may find it difficult to interpret ICH guidelines, understand regional regulatory requirements, or use complex publishing tools. This often leads to delays, subpar submissions, and repeated corrections. The training process for new regulatory professionals takes a long time because they need both scientific knowledge and technical publishing skills. The lack of skilled personnel particularly impacts smaller companies, where regulatory tasks often fall to individuals with limited expertise. In general, human resource limitations greatly affect the efficiency and accuracy of CTD/eCTD preparation.
Data Integrity And Quality Submission Delays:
Data integrity is one of the most important expectations from regulatory authorities. All data submitted in CTD/eCTD must be complete, accurate, current, and traceable. However, keeping high standards of data integrity across multiple departments can be tough. Inconsistent record-keeping, manual documentation practices, insufficient audit trails, outdated data, and poor version control create gaps in data integrity. Sometimes, multiple versions of the same report circulate internally, causing confusion about which is the final version.Quality issues such as missing Certificates of Analysis (COAs), incomplete stability summaries, or differences between analytical methods in various studies can slow down the submission process. These problems force teams to redo reports, conduct additional checks, or update multiple modules at the last moment. As a result, the timelines for compiling dossiers are extended, delaying the overall submission.
AI TOOLS USED IN CTD AND ECTD PREPARATION
AI is increasingly playing a significant role in regulatory submissions. Peer-reviewed research highlights AI as a game-changing technology for automating regulatory documents, predicting metadata, and managing lifecycles.
Major AI tools used in CTD and eCTD:
Lorenz DocuBridge AI:
This is one of the leading intelligent publishing platforms used by many regulatory authorities and pharmaceutical companies around the world. Its AI features are designed to automate time-consuming tasks, such as classifying documents, managing lifecycle operations, and validating XML structures. The system learns from past submissions and assists regulatory teams in automatically identifying the correct module placement for each document. It can spot incorrect lifecycle operators, recognize metadata patterns, and validate the XML framework against regional specifications. This lowers the risk of technical rejections and improves compliance with ICH M8 requirements. DocuBridge's AI engine also helps with preparing submission sequences more efficiently by analyzing document relationships, predicting updates, and cutting down on the need for manual tagging or organization. As a result, companies benefit from a more consistent and accurate publishing process across various global markets.
EXTEDO's eCTD Manager AI:
This has also become a widely used solution for intelligent regulatory publishing. It is noted for its ability to automate hyperlink creation and predict metadata for documents within the CTD framework. Hyperlinking, which traditionally takes a lot of manual effort, is generated automatically by the system based on the document's content and past submission patterns, ensuring proper navigation throughout the dossier. The AI also helps predict leaf titles and metadata fields, thus reducing mistakes in manual data entry. This enhances navigation for reviewers and supports compliance with technical formatting guidelines. EXTEDO's AI features also improve quality control by spotting missing bookmarks, broken links, wrong PDF versions, or misplaced documents, significantly lowering the risk of validation failures during agency submissions.
Veeva Vault RIM:
This system, which integrates artificial intelligence, is considered one of the most advanced global regulatory information management systems. Its AI layer enhances submission planning by predicting regulatory requirements for different markets, analyzing regional guidelines, and offering smart recommendations for submission timelines and document readiness. Veeva's AI also helps with version control, automated metadata tagging, and compliance monitoring by identifying outdated or inconsistent documents. It keeps a centralized and harmonized regulatory repository that allows global teams to work together efficiently. The predictive capabilities assist organizations in anticipating regulatory questions, managing lifecycle updates, and optimizing submission planning across various regions. The combination of RIM and AI ensures that data integrity, submission documents, and timelines remain coordinated throughout the entire regulatory lifecycle, cutting down on delays and strengthening regulatory strategy.
Ennov Regulatory AI Suite:
This tool aids regulatory operations by focusing on workflow automation and compliance tracking. Unlike traditional publishing tools, Ennov's AI focuses on process optimization, helping organizations manage submission planning, review cycles, task assignment, and document readiness. The system tracks the progress of each submission component, identifies delays or issues, and sends automated alerts to stakeholders. It looks at patterns in submission workflows and suggests improvements to reduce bottlenecks. Ennov's AI also promotes compliance by making sure that standard operating procedures, templates, and processes used in preparing documents meet the latest regulatory requirements. This helps regulatory affairs teams maintain a high level of document quality and consistency in operations, especially when handling multiple submissions at the same time.
Freyr SUBMIT PRO AI Engine:
This tool has emerged as a strong and intelligent regulatory publishing and management ecosystem. According to industry reports, its AI features are specifically designed to perform gap analysis, identify missing documents, and align content with global submission templates. The system interprets regulatory guidelines and compares current dossiers with specific regional requirements, enabling companies to prepare submission-ready documents more easily. Freyr's AI engine also helps harmonize data across modules, highlighting inconsistencies and suggesting corrective measures to ensure scientific alignment throughout the dossier. Its ability to recommend global template structures makes it especially useful for multinational submissions, where different agencies may require variations of the same document. Through its predictive insights and automated processing, Freyr SUBMIT PRO helps regulatory teams minimize errors, enhance submission completeness, and speed up publication timelines.
FUTURE TRENDS IN GLOBAL REGULATORY SUBMISSIONS
Future regulatory changes will focus on further digitalization. The articles show several trends emerging globally.
eCTD V4.0 (HL7 RPS):
eCTD v4.0 allows two-way structured messaging between regulators and the industry. Studies indicate it brings improved detail, better metadata standardization, and smoother lifecycle transitions 12-13.
Cloud-Based Submissions:
Cloud systems enable real-time editing, integrated validation, and collaborative workflows. Industry trend reports identify cloud regulatory information management (RIM) systems as the future standard 11.
Blockchain For Data Integrity:
Blockchain may soon be used to prevent data tampering and provide secure audit trails.
Digital Labeling (E-Labeling):
The globally accepted digital labeling allows quick updates and reduces errors. Both the EMA and FDA back e-labeling initiatives 9-4.
RESULTS AND DISCUSSION
The review demonstrates that CTD and eCTD have significantly improved global regulatory submissions by ensuring standardization, reducing duplication, and enabling simultaneous submissions across regions. The transition to eCTD introduced digital features like XML backbone, lifecycle management, and automated validation, enhancing review efficiency and transparency. Widespread adoption by major regulatory agencies reflects strong global harmonization, although regional differences still exist. Challenges such as technical errors, high costs, data fragmentation, and limited skilled workforce remain concerns. The integration of artificial intelligence has improved accuracy, reduced manual effort, and minimized errors. Future advancements like eCTD v4.0, cloud systems, and digital technologies will further streamline regulatory processes.
CONCLUSION
The CTD and eCTD remain essential parts of global drug regulation. Supported by efforts for harmonization, digital transformation, and emerging AI technologies, they continue to strengthen the reliability, quality, and speed of drug evaluation worldwide, ultimately benefiting patients and the global healthcare system.
The Common Technical Document (CTD) and its electronic version (eCTD) have transformed global regulatory submissions by introducing uniformity, efficiency, and transparency into the drug approval process. The CTD ensures consistent documentation across ICH regions, while the eCTD further enhances this process through digitalization, automated navigation, lifecycle management, and reduced review times. As pharmaceutical development becomes more complex, the eCTD serves as a robust platform for real-time updates, secure data transfer, and improved regulatory communication. Additionally, the integration of modern digital tools and artificial intelligence is further advancing dossier preparation, validation, and submission accuracy. Overall, the CTD and eCTD standards continue to play a crucial role in speeding up patient access to safe and effective medicines around the globe.
REFERENCES


Shrutika Dagade, Bharatee Chaudhari, Global Harmonization of Common Technical Document and Electronic Common Technical Document, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 4, 1191-1202 https://doi.org/10.5281/zenodo.19466229
 10.5281/zenodo.19466229
10.5281/zenodo.19466229
