1M. pharm, Department of Pharma Regulatory Affairs, K.B. Institute of Pharmaceutical Education and Research, a constituent college of Kadi Sarva Vishwavidyalaya, Gandhinagar.
2Industrial guide of Makcur Laboratories Ltd, Ahmedabad.
3Head of Department of Pharma Regulatory Affairs, K.B. Institute of Pharmaceutical Education and Research, a constituent college of Kadi Sarva Vishwavidyalaya, Gandhinagar.
4Assistant Professor of Pharma Regulatory Affairs, K.B. Institute of Pharmaceutical Education and Research, a constituent college of Kadi Sarva Vishwavidyalaya, Gandhinagar.
The pharmaceutical regulatory frameworks in Kenya and Saudi Arabia ensure drug safety, efficacy, and quality. Understanding their registration processes is crucial for pharmaceutical companies seeking market entry. This study compares the drug registration requirements of both countries, focusing on regulatory authorities, dossier submission, approval timelines, and key challenges. A comprehensive review of regulatory guidelines from the Pharmacy and Poisons Board (PPB) of Kenya and the Saudi Food and Drug Authority (SFDA) was conducted. Although both countries align with ICH and WHO standards, differences exist in Common Technical Document (CTD) vs. eCTD submission formats, local testing requirements, and approval timelines.
Regulations, semi -regulation, and registration of pharmaceutical products in other countries in the world are difficult procedures. In regulated nations, the criteria are in line with CTD (common technical document) filing; however, in other countries, the standards vary wildly. Pharmaceuticals and regulators in various countries, In various aspects of drug registration, including the United States, Japan and Europe, we participate in ICH I (International Conference on Harmony of Technology Requirements for Pharmaceuticals for Human Use). Similarly, the Persian Bay Cooperation Council (GCC) and the Southeast Asian National Association (ASEAN) are striving to harmonize the general problems of the Asia - Pacific region. Requirements must be optimized, and this may be assessed by looking at the prevalence of increased costs associated with the availability of medications and R&D facilities. It must be a medicine It has been justified and rationalized for public safety to improve treatment safety, efficiency.2 The duties of the regulatory authorities are as follows; to standardize, monitor, oversee, control and approve all the existing and forming medicines in their country to guarantee their safety, quality, and efficacy. Making sure that pharmaceutical products are developed in compliance with the national regulatory standards is one of the main challenges facing the regulatory agencies.
Introduction of Drug Registration Process
Drug registration is an important final step of drug evaluation, before they are marketed and made available to the public as a safe and effective form of treatment of a particular disease. Various governments of the world’s sovereign countries demand that drug manufacturers provide a vast amount of information on their products before they can be allowed to market them. The process is intended to reduce distribution of poor quality, unsafe or ineffective products that compromise public health.5
Global Drug Registration Landscape
The healthcare regulatory authorities for drug registration differ from one country to another, and complete sets of rules and regulations exist for each area. Nevertheless, the fundamental parameters of the drug registration procedures are generally similar in all countries.
International Regulatory Bodies and Guidelines
Several international regulatory bodies and agreements influence drug registration globally:
World Health Organization (WHO): These guidelines involve; Good Manufacturing Practices (GMP), Good Clinical Practices (GCP) as well as WHO recommendations on the quality and safety of Drugs. It also enables facilitating drug registration in the prequalified countries especially those in the least developed countries.
International Council for Harmonization (ICH): ICH is conference of regulatory authorities of United States, Europe and Japan with objective to ease drug development and registration in different countries based on quality, safety and efficacy criteria. The ICH guidelines are considered authoritative worldwide and function as the basis for the standards used in a number of states.
Pharmacopeias (USP, EP, and BP): These are collections of standards of drugs. Some areas may use particular pharmacopoeias, for instance, USP, EP or BP that contain desk specifications for medical ingredients and dosage forms.
General Format Of CTD:
The information included in the CTD should be clear and unambiguous, just like in any other document. As per the ICH M 4 guidelines on the format of CTD (USA), text and tables are to be formatted with margins to enable the document to be typeset both A4 (which is utilized in the EU & Japan) and 8.5/11 inch paper. Narrative prose should be written using times new roman with a 12 point font size. Acronyms and abbreviations in any section of the learning module must be defined the first time they occur in that module for meeting the URMSM-BJ requirements. Additionally, the literature cited by the learning module should be referred to at the end of the module. (Source-ICH M4 Guideline) 6.
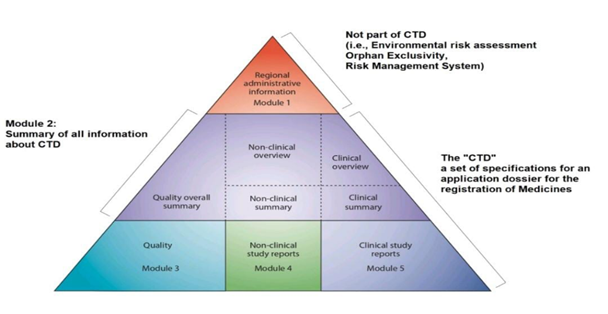
The CTD documents are divided into five parts (see figure 1):
Module 1: Administrative & prescribing information.
Module 2: Module 3 to 5 Overviews and Summaries
Module 3: Pharmaceutical documents (Quality)
Module 4: Reports on Non-clinical study (Pharmacology/Toxicology)
Module 5: Reports on Clinical study (Clinical Trial)
Electronic Common Technical Document E-CTD:
ICH M2 EWG has installed electronic general technology documents as an electronic framework for submitting regulatory data related to the creation, review, and life cycle, the ICH M4 EWG created the form of the so-called Common Technical Document. In addition to open standards, the eCTD is tailored for regional submissions and adds regional elements (module 1), further expanding the versatility of the CTD (modules 2–5). (Source-ICH M8 Guidelines) 6.
Pharmaceutical Market:
Pharmaceutical market is segregated into following categories: 3
1. Regulated market: these are america, european union (uk, germany, france, ireland and sweden and many others.), japan, canada, australia, new zealand, and south africa
2. Semi regulated market:
a) Asia: The countries of SAARC are sri-lanka, india, bangladesh, china, pakistan, bhutan, and nepal
b) Asean: ASEAN 10 international locations are the philippines, vietnam singapore, malaysia, thailand, indonesia, laos, cambodia, brunei darussalam, and myanmar
c) African countries: The look at become accomplished among college students of chosen universities in Anglophone nations in Africa, inclusive of algeria, zambia, ethiopia, ghana, kenya, malawi, mozambique, namibia, nigeria, sierra leone, tanzania, zimbabwe
d) Middle east nations: deciding on countries which might be individuals of the Gulf Co-operative Council structures, that is bahrain, kuwait oman, qatar, saudi arabia and the UAE
e) Latin the usa: (mexico, brazil, panama, peru, guatemala, argentina, chile, dominican republic)
f) CIS (common wealth of independent states) : russia, ukraine, post-soviet states (armenia, azerbaijan, belarus, georgia, kazakhstan, kirghizstan, moldova, tajikistan, turkmenistan, and uzbekistan
Regulatory Agencies
Regulatory bodies and agencies are very important in making sure that a country discharges its legal requirement in the creation of Pharmaceuticals. Each nation has its own regulatory bodies that are tasked with the enforcement of the laws and regulations and establishing conditions for manufacture, approval, distribution and registration of drugs. Ensuring the efficacy, safety & quality of drugs involves provision of support to various initiatives which is one aspect of drug regulation.3
Introduction Of Kenya And Saudi Arabia
Eastern African Countries:
In eight (8) partners, the Local government organizes the East African Community (EAC). Somali Federation, Southern Sudan Republic, Burundi Republic, Congo Democratic Republic, Kenya Republic, Rwanda, Uganda, Uganda Republic and Tanzania Republic are the headquarters of the Republic of Arusa and Tanzania 13. On November 24, 2023, the Somalia Federal Republic entered into the East African Community by EAC summit of heads of state. After that, the Federal Republic of Somalia joined EAC by signing the treaty of accession to the EAC treaty on 15 december 2023. The federal republic of somalia becomes a full member immediately after she deposited her instrument of ratification of the EAC treaty with the EAC secretary general on 4 march 2024.
Kenya:
Market:
Current pharma market is $ 9.4 billion and it is expected that by the end of next five five years, with the compound growth rate of 85%, will be over $ 1.4 billion. Global comparisons show that the per capita drug expenditure is actually very low; but, then again, this is relative to the general trend of the other members of the Sub-Saharan Africa region, such as Nigeria. Many individuals in Kenya belong to the lower end of the spending potential scale. This is inevitable that the demand for cheap pharmaceuticals will prevail, so the possibility of a multinational pharmaceutical company is inevitable. But this is an opportunity for ordinary exporters in India. 13
Generic Drug market:
Increased volume consumption, resulting from demand and support of measures, which seek to make universal health coverage, shall be the key drivers for the growth of the generic medicines industry in Kenya. Additionally, limited per capita purchasing power will further favor less expensive items, even while pricing research exposes quality issues in local generics producers. Out of total imports in formulation, almost all are re-exported from India. Generic market the was $ 610 million in 2017 and it comprised 63.9% of the entire market. Analysts predict an increase of 10% in this year. 13
Pharmaceutical Trade:
More refined treatments are imported to fill most demand within its borders despite the fact that most locally manufactured drugs are intended for the domestic market. In the forthcoming years increasing demand in the given sector, in particular, in pharmaceuticals, is going to additionally worsen the trade balance as the majority of the domestic producers do not have necessary funds for innovations and upgrades. On a cumulative basis, the overall import/export statistics remain distorted by its function as a regional trading center. 13 Like most emerging economy, Kenya remain to exhibit a trade deficit as indicated in the analyst. Drugs and medicines imports are $648mn in 2017 and is projected to reach $728mn in the year ended during December 2018. Export, which the country realized in a small amount of USD102 million in 2017 will increase to USD143 million.
Regulatory Review:
Pharmaceutical and medical device registration in Kenya is handled by the Pharmacy and Poisons Board (PPB), which was founded in accordance with Chapter 244 of the Pharmacy and Poisons Act (2002). Pharmaceutical testing for regulatory purposes is handled by the National Quality Control Laboratory. In practice, nevertheless, it is thought to test fewer than 20% of samples. It is expected that importers will comply with legal requirements, including sending drug samples to the Kenya Bureau of Standards for registration and quality control and following national policy directives set forth by the Ministry of Health. Using WHO recommendations, this is a list of important pharmaceuticals with the goal of promoting the accessibility of high-quality pharmaceutical products at reasonable costs. 13
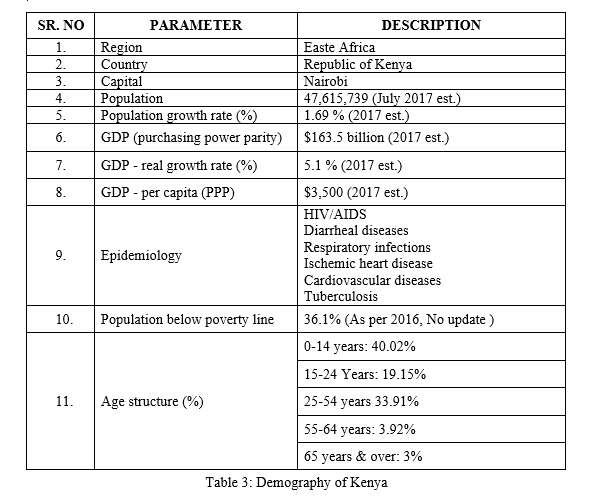
Demography:
Gulf Co-Operation Council (GCC) Countries:
The gulf cooperation council (GCC) is also said to be the cooperation council for the arab states of the gulf (CCASG). This is the political and economic union of the Arabian Peninsula, a collection of Middle Eastern states bordering the Persian Gulf. Also commonly referred to as "The GCC States," the 6 member states of the union are Oman, Saudi Arabia, Qatar, Kuwait, Bahrain & the United Arab Emirates. On 11/11/1981, in Abu Dhabi, these Gulf Cooperation Council countries signed their collective economic agreements 16. Saudi Arabia is one of the most attractive Middle Eastern markets for multinational corporations. The diversification ambitions of the Kingdom are in the interest of regional and domestic companies since "Vision 2030" favors the growth of manufacturing deals within the country. Multinational corporations are keenly interested in Important factors are the size of the market, the demand's complexity, and the positive epidemiological trends. As demand for the established general procedure increases, there will be a successful commercial base for local players. However, for international research-based pharmaceutical companies, problems with patent approvals and the regulatory framework continue to be a significant concern. With a 5.2% annual growth rate, the market is predicted to reach USD 8.8 billion by the last of 2020 from its current value of USD 8.3 billion in 2019.
Pharmaceutical Market for Saudi Arabia:
The pharmaceutical market of Saudi Arabia was worth USD 8.3 billion in 2019. In 2024, the market with an annual growth rate of 5.4%will reach $ 10.8 billion. USA. Populence and consumers' and prescribers' craving for branded medicine enhance the sale of patented consumption of drug that covers 54.4% of the market of the country. Even though it has the largest regional pharmaceutical market by total value, it is seventh in the region by drug expenditure per capita, at USD 242 16. Saudi hospitals are the best in the middle east’s best, but the country's specialized tertiary institutions are on par with those in Western Europe. There are extensive plans for the construction of new hospitals and health facilities, among other infrastructural improvements. According to the WHO, non-communicable illnesses account for 84% of Saudi Arabia's fatalities, which is consistent with the nation's known epidemiological profile. According to mortality, the three most common chronic diseases are strokes (16%), ischemic heart disease (24%), and cardiovascular disorders (49%).
Generic Market
The government's encouragement of generic substitution as a cost-saving initiative promotes the Saudi Arabian generic drug market and is a key growth driver for this phenomenon. Even if the kingdom's regulatory standards have improved somewhat, industry favoritism of domestic producers will help Saudi Arabia's generic medication market, which is expected to expand quickly in medium and long-term as compared to its patented counterparts 16. The industry is predicted to growth at an annual rate of 8.1% over the following 5 years, from its current size of $2.9 billion (which represented 35% of the market in 2019) to $4.4 billion by 2024, when it would account for 39% of the market. Encouragement from the government drives the nation's local generic business. Attempts (primarily in the public sector) to get the region away from its over-reliance on imported drugs will be supported by intentions for more regional integration and more of a focus on GCC-made pharmaceuticals.
Pharmaceuticals Trade
The domestic drug industry of the Kingdom will thrive through the assistance of Saudi Arabia's International Transformation Plan and Vision 2030. This will lessen the need for imported generic medications while opening up export markets to nearby import-dependent states. In 2019, the nation imported $5.9 billion. With a 4.4% CAGR, imports are expected to reach $6.6 billion by 2023. Saudi Arabian companies, primarily the government purchasing agency NUPCO and the private sector Banaja Saudi Import Company, are solely in charge of import distribution. Greater collective bargaining among GCC nations, regional harmonisation, and local bias in favour of local and regional producers are anticipated to support local production and increase exports 16.
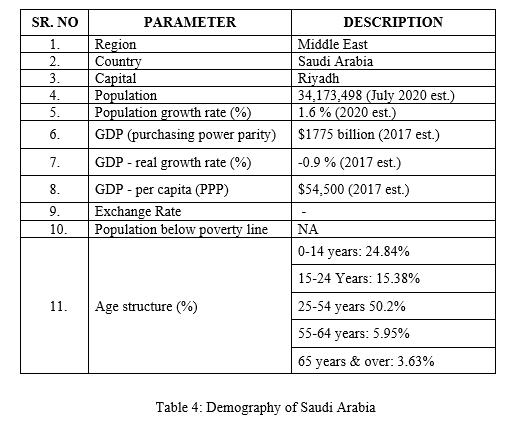
Demography
METHODOLOGY
Kenya
Regulatory authority of Kenya
Pharmacy & poisons board: The Pharmacy and poisons board, sometimes known as the Board or PPB, was created the Drug Regulatory Authority as per the provisions of the pharmacy & Poisons Act, Chapter 244 of the Laws of Kenya.
The board is in charge of regulating the manufacture and distribution of drugs and poisons, as well as the practice of pharmacy 14.
Requirements For Registration: [13]
1. One local agent with a general power of attorney is required for every overseas producer. The local agent needs to be a licensed Kenyan drug wholesaler.
2. Free sales certificate or medical product certificate of origin.
3. A distinct application for every item.
4. An MS-Word document should encompass Modules 1 and 2 in a cross-reference manner to the dossier by clearly stating the title and section number of all the supporting papers, and be submitted in one original hard copy and one electronic copy (in a Portable Document Format, PDF, or on a CD-ROM).
5. GMP compliance is required of the manufacturer. The Board retains the authority, at the applicant's expense, to confirm the manufacturer's compliance with good manufacturing practices.
6. Three (3) samples with batch certificates of analysis of the smallest commercial pack or packs from a single batch.
7. Certificate of Pharmaceutical Product with authentic WHO Format issuing from the competent authority of the country.
8. If the master file site product is studied by PPB and is not yet approved by an object or factory.
9. It is impossible to submit an application for drug registration to the GMP inspection fee ($ 6,000) and Kenya to study and approve the PPB.
Step by step procedure:
Perform the following steps in sequence:
1. Go to the Kenya Drug Registration system.
•The website is http://www.pharmacyboardkenya.org.
2. Login so that you can apply (every applicant should have a username and password2).
3. Select the appropriate module for Medicinal Product Registration.
4. Product dossier Submission
5. PPB conduct pre-screening to ensure submission is complete.
6. Pay the applicable registration fees through to the portal.
7. Inspection and Laboratory Testing.
8. Approval and Issuance of registration Certificate.
The next step is to approve the drug for all new pharmaceutical products, including things obtained from biotechnology.
- Getting applications
- Authorization of the market agency
- Inspection of manufacturers and production facilities for adherence to modern GMPs
- Analysis of international quality management laboratory
- Recommended for registration of drugs
- Working Committee Review Committee
- Complete consent of the board of directors
- Newspaper
If questions are not sufficiently addressed within six months of the request, the drug application is deemed withdrawn. If a medicine is denied, the applicant has two months from the notice date to contest the decision.
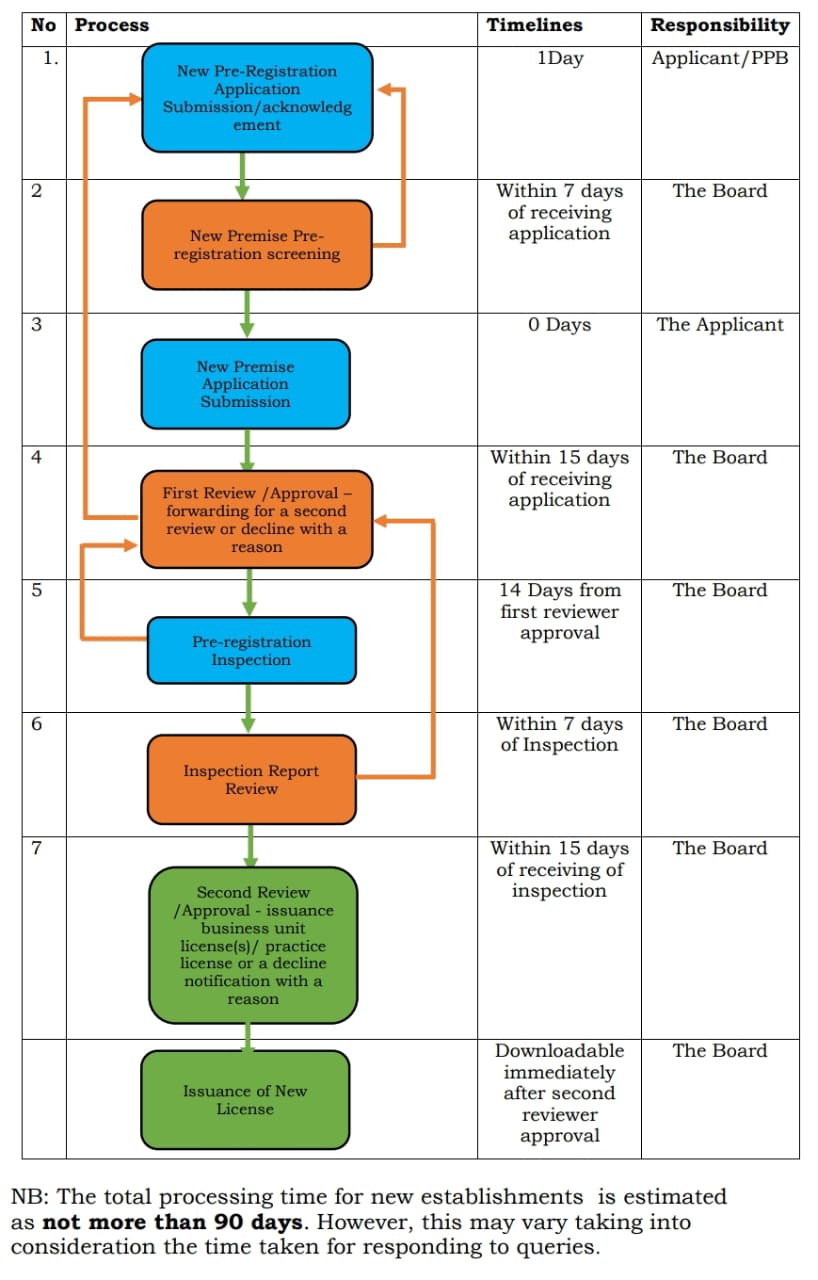
Fast Track registration: [13]
It is possible to expedite an application if the product is
- Produced locally in Kenya.
- The recommendation of the product would pertain to the class of illnesses with no approved alternative medication, or there is evidence that it offers obvious advantages over existing products in terms of safety and efficacy for treating life-threatening anomalies. Completed applications, including documentation review and committee consideration for drug registration, will be handled within 90 working days of receipt.

Validity of Registration:
A pharmaceutical product's registration is good for five (5) years unless the PPB suspends or revokes it beforehand or the applicant withdraws it.
Fee Structure:
For international producers looking to ship medications to Kenya [13]

Language:
Every application and accompanying document needs to be in English and readable.
Application Submittal
Application grouped into three categories for submission to PPB are as follows:
(1) New application form
This will be sold for the first time in Kenya as a drug registration application. Each product requires its own application. Even if they contain the same chemicals, goods with different active ingredient(s), strengths, dosage forms, and proprietary names are regarded as distinct products and, as such, need various applications [15]. The same standard and dose type, according to the form of dosage, the same product is similar to the active ingredient and the products manufactured are different from the size of the packaging or packaging, so it should be displayed as one application.
A new registration application must contain:
(i) Module 1 and 2 and confirmation documents containing CD-ROM; Two types of applications that are completed correctly, original and replication; And an electronic copy of the document outline of the Microsoft word format.
(ii) Sample: Package certificates of the smallest commercial package or three samples packed in one batch.
(iii) In the initial document issued by the legal certification of the WHO -form pharmaceutical product, the drug regulatory authority.
(iv) Master file of the site produced on the site is not confirmed and approved by PPB.
(v) Invincible fees for paying GMP inspections and Kenya's drug registration applications in an institution that has not yet been investigated by the PPB.
(2) Applications for Renewal of Registration
The following is required to be provided with an application for a registration renewal three months before the original registration runs out:
i. Completed application for a registration renewal.
ii. Batch Manufacturing Record (BMR) of an actual batch produced no more than six months prior to application submission.
iii.Periodic Safety Update Reports (PSUR) are to be provided.
iv.Documentation to prove that the generics can interchange.
v. Any additional details as deemed appropriate by the Board.
vi. Three Package certificate, the smallest commercial package or sample supported with the same batch package.
vii. Master files of the site if the product is manufactured in a facility or factory that has not been visited or not approved by PPB.
viii. Visit and non -incredible fees for Kenya Pharmaceutical Registration and GMP inspection registration application without visits and approved by the PPB and GMP departments.[15]
(3) Application for variation of registered medicinal product
All requests for the registered changes of the product must be observed according to the changed medicine change according to the PPB management manual. If you are submitted after the reliability period of the registration certificate, it is considered a new statement of form 1. [15]
Evaluation process
Unless the product satisfies the fast track requirements outlined in this guideline, applications are evaluated on a first in, first out (FIFO) basis. Fast-tracking an application is possible if the product is:
- Produced locally in Kenya. It should be noted that a Kenyan company's contract manufacturing outside of Kenya does not equate to local manufacturing.
- A priority medicine is one that is prescribed for conditions for which there is currently no approved alternative medication or for which there is proof that the product offers notable safety and effectiveness benefits over currently available medications for treatment or prevention of life - threatening conditions. Evaluators from PPB or outside the company evaluate product dossiers. A second evaluator reviews the evaluation report that the first one created, performs quality assurance on the report, adds comments as needed, and completes the report and recommendations. In accordance with the standard operating procedures for assessment, the assessment is carried out in relation to the requirements of the guidelines. The Board does, however, retain the power to ask for any more data necessary to determine a medication's efficacy, safety, and quality in light of the state of knowledge at the time of evaluation. A query letter may be used to request further data and/or samples during examination. The procedure ends when a question is posed and sent to the applicant, and it continues until PPB receives a written answer. Only when answers to the questions posed in the same letter are sent in as a single transaction for review can the application be concluded. The product will be excluded and the application will be refused if this requirement is not met or if the questions have been reprinted twice and the applicant gives inadequate answers. If the responses are not submitted within six months after the questions' issuance, the applicant will be deemed to have withdrawn the application. After that, product registration might only be taken into consideration when a fresh application is submitted [15].
Timelines
When processing applications for pharmaceutical product marketing permission, the Board will adhere to the following schedules. Accelerated registration (only for locally produce and Priority medications), post-approval variation, and registration renewal.
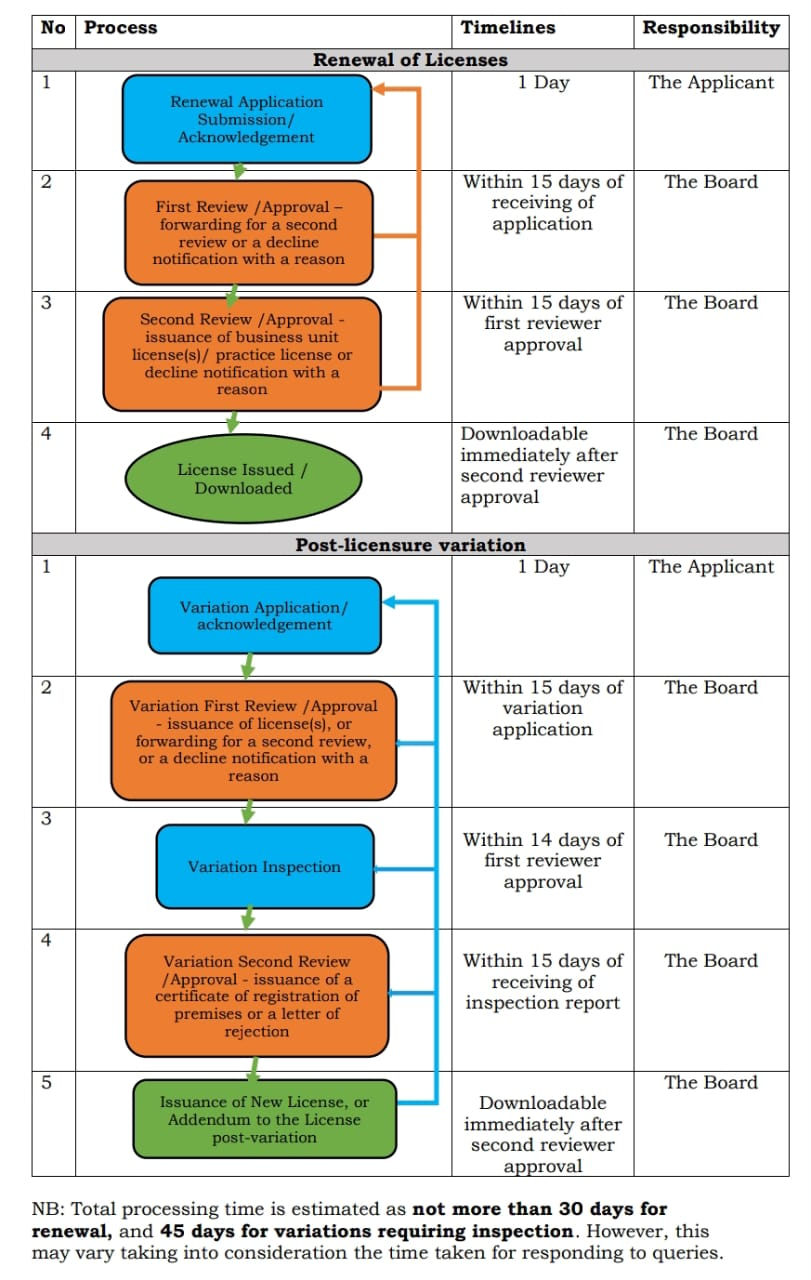
Renewal and Variation Timeline Process
Completed applications, including documentation review and committee consideration for drug registration, will be handled within 90 working days of receipt.
Validity of Registration
A pharmaceutical product's registration is good for five (5) years unless the PPB suspends or revokes it beforehand or the applicant withdraws it. When the Board suspends, revokes, or modifies registration conditions, it will provide written justification. Similarly, the applicant must provide justification for canceling a product's registration [13].
Appeals
Anybody who feels wronged by a PPB decision regarding a pharmaceutical product's application for marketing authorization has two months from the date of decision notice to submit written comments to the Board, pay the required appeal cost & provide more information to back up their claims [13].
Saudi Arabia
Regulatory Authority of Saudi Arabia
Saudi food and drug authority
The Saudi Food and Drug (SFDA) was created as an independent enterprise organization that informs the chairman of the Ministers' Council according to the Ministerial Council. Resolution number (1) 07/01/1424 H. It is a force to ensure the safety of food and drugs for biological and chemicals, electronic devices, foods and animals. [16]
Authority Activities:
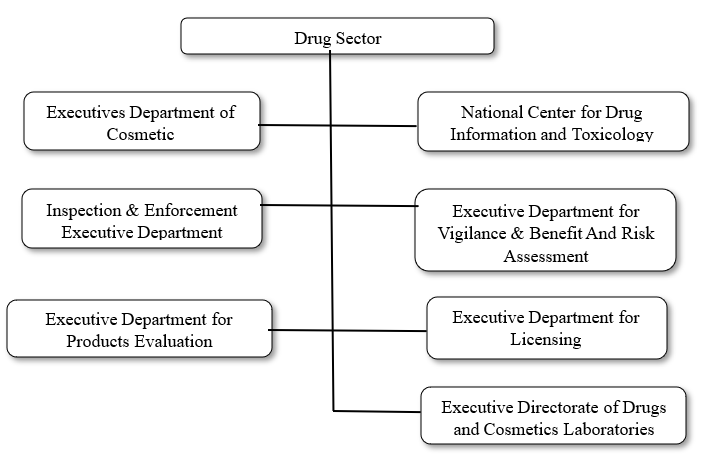
Figure 9: Authority Activity
Requirements For Registration:
• Information must be configured according to the recommendations of the electronic general technology document (e-CTD)
• All documents should be readable; they must always be text searchable and Tables included, the page size must be consistent.
• Licenses and letters of approval, among other supporting documentation, must be in Arabic or English.
• A document has to be translated into English and stamped by the Saudi Embassy from the origin country if the document is neither in Arabic nor in English.
• The language on the label must adhere to the SPC (Summary of Product Characteristics) requirements; the Saudi Arabian government must approve the labeling since it is an essential part of the product's license [16].
Process Of Submission [17]
The three stages are the process of supplying drugs in SFDA.
1. Submitting an online application form
1. Send online forms for submitting applications 2. Provide hard copy and soft copy of product files
3. Pharmaceutical sample
Opinion:
• Hard copy (driver) must be followed by the format of the general technical document (CTD).
• See Soft Copy (Computer Copy -CD or DVD) to receive additional information "File Manual".
• All days mentioned in this document are working days.
• All days referred to in this document are WORKING days (tentative).
Step by step procedure:
1. Applicant visit the website SFDA.
2. Login for use (each applicant must have a user identifier and password2.)
3. Select the appropriate application and write it.
• The application can be filled in several stages, so the application can be preserved in part.
4. Then, in order to send an application form and organize the hard and soft copy of the product file, the applicant must pay a fee for submitting (through the Sadad Payment System or by Bank Deposit).
• You need to eat to move to the next stage.
• The fastest purpose can be done 1-12 weeks before. Candidates can transfer a week before the meeting. An automatic notification is provided three days before the appointment.
• The link number is provided and you must always use the link number in relation to communication with SFDA.
5. During the meeting, the applicant provides product files (hard copies and soft copies) and samples.
6. The staff in the pharmaceutical sector will be certified as follows:
a. Application form
b. Product document (Hard & Soft copy)
c. Samples
• An acknowledgement letter will be created and provided to the applicant if the aforementioned are confirmed. The drug application will enter the queue.
• An acknowledgment letter outlining the shortcomings will be created and provided to the applicant if any of the aforementioned are lacking or unsatisfactory. The drug application will not be placed on the queue, and the applicant will have sixty days to meet the requirements. If not, the product file will be safely disposed away by SFDA.

Flow chart of Submission Process
Fee Structure
Fees for Pharmaceutical Product Regulatory Services [16]
ESULT & DISCUSSION
Here, I made by dossier from my oun side as per regulatory guidelines as per regulation of Kenya & Saudi arabia. Its Summary is included in differentiate and simmilarities of my dossier in chart table form.
Demographic Structure:
|
Parameters |
Kenya |
Saudi Arabia |
|
Regulatory Agency |
Pharmacy and Poisons Board (PPB) |
Saudi Food And Drug Authority (SFDA) |
|
Country Flag |

|

|
|
Member |
East African Community (EAC). |
Gulf Cooperation Council (GCC) |
|
Surface Area |
580,370 km square |
2,149,690 km square |
|
Capital |
Nairobi |
Riyadh |
|
Population |
52,428,290 |
34,173,498 |
|
Population growth rate (%) |
1.69 % |
1.6 % |
|
GDP (purchasing power parity) |
$338.964 billion |
$1,775 billion |
|
GDP - real growth rate (%) |
5.1 % |
2.6 % |
|
GDP - per capita (PPP) |
$6,576 |
$ 54,500 |
|
Population below poverty line |
63 % |
NA |
|
Dossier Format |
CTD |
eCTD |
|
Fees for Drug Registration |
USD 1000 |
SAR 40,000 |
|
Time taken for registration |
12 Months |
6 - 18 Months |
Registration Fees:
|
|
Kenya |
Saudi Arabia |
|
Fees by |
US$ |
Saudi Riyal |
|
Application for the Registration of a Drug product |
US$ 1,000 |
40,000 |
|
Fee for GMP Inspection of Foreign facility* |
US$ 6,000 |
10,000 |
|
Appeal fee |
US$ 300 |
20,000 |
|
Variation Fee |
US$ 200 |
3000 |
|
Replacement of a registration Certificate |
US$ 100 |
33,000 |
|
Annual Retention fee per product |
US$ 300 |
- |
|
Renewal of Registration certificate |
US$ 500 |
30,000 |
|
Fine for late renewal |
US$ 500 |
- |
|
Evaluating the addition of a new pack type |
- |
24,000 |
|
Evaluating the addition of a new pack size |
- |
1,000 |
Responsibilities of the regulatory authorities:
|
|
Kenya |
Saudi Arabia |
|
Medicines for human use |
√ |
√ |
|
Veterinary Medicines |
√ |
√ |
|
Medical Devices and In-Vitro diagnostic |
√ |
√ |
|
Cosmetic Products |
× |
√ |
|
Food Supplements |
× |
√ |
|
Herbal Medicine |
× |
√ |
|
Scope of activity Marketing Authorization |
√ |
√ |
|
Post-marketing surveillance |
√ |
√ |
|
Sample analysis |
√ |
√ |
|
Advertising Control |
√ |
√ |
|
Price regulation |
√ |
√ |
|
GMP inspection |
√ |
√ |
|
Clinical trial authorization |
√ |
√ |
Standard Estimation and Aspect
|
|
Kenya |
Saudi Arabia |
|
Verification Review |
× |
√ |
|
Abridged Review |
√ |
× |
|
Full Review |
√ |
× |
|
Fast track time (Calendar Days) |
90 days |
NA |
|
Variation requiring inspection |
45 days |
30 days |
|
Evaluation of documents & consideration drug registration |
90 days |
90 days |
|
Evaluation of new application |
12 months |
NA |
|
Withdrawal of an application |
6 months |
NA |
|
Appeals pharmaceutical product |
2 months |
60 days |
|
Assessment Report |
NA |
90 days |
|
Pricing |
NA |
90 days |
Enclosures required for drug registration in Kenya and Saudi Arabia
|
Sr. No |
|
Kenya |
Saudi Arabia |
|
1. |
Control Specific and method of analysis |
R |
R |
|
2. |
Certificate of analysis attested by health authority and country of origin |
R |
R |
|
3. |
Legalized free sale certificate issued by health authorities for coo, indicating that product registered and marketed with same name & composition |
NR |
R |
|
4. |
Legalized certificate indicating that diluents used are allowed to be in certificate of origin (COO) |
NR |
R |
|
5. |
Legalized price certificate issued by component authority of coo &attested by embassy including-factory price, wholesale price COO |
R |
R |
|
6. |
Retail/Public Price in COO |
R |
R |
|
7. |
EXPORT price to country and neighboring countries |
R |
R |
|
8. |
Stability studies in various defined conditions |
R |
R |
|
9. |
Storage conditions |
R |
R |
|
10. |
Name of developed countries in which the product is registered |
R |
R |
|
11. |
Abstract from scientific references about product |
R |
R |
|
12. |
Sealed sample of product and copies of label |
R |
R |
|
13. |
Quantity specified for each pack and outer pack of product |
R |
R |
|
14. |
Leaflet in Arabic and English, including |
||
|
|
|
English |
Arabic & English |
|
A |
Name of Product |
R |
R |
|
B |
Composition |
R |
R |
|
C |
Mode of action |
R |
R |
|
D |
Effect |
R |
R |
|
E |
Indications |
R |
R |
|
F |
Contraindications |
R |
R |
|
G |
Precautions |
R |
R |
|
H |
ADR |
R |
R |
|
I |
Antidote |
|
R |
|
J |
Dosage and Administartion |
R |
R |
|
K |
Storage |
R |
R |
|
15. |
Product Labeling |
R |
R |
|
16. |
Bioavailability study |
|
R |
|
17. |
Scientific basis of justifying the formulation of combination product |
R |
R |
|
18. |
Post marketing surveillance |
R |
R |
|
19. |
Certificate of sterility and pyrogen free pharmaceutical product |
NR |
NR |
|
20. |
Copy of reference pharmacopoeia |
R |
R |
|
21. |
CTD format for dossier submission |
R |
R |
|
22. |
e CTD |
NR |
R |
Note:
R: Recommended by regulatory authority
NR: Generally, not recommended by regulatory authority
Submission Requirement
|
|
Kenya |
Saudi Arabia |
|
Format |
CTD format |
CTD format. e-CTD recommended. |
|
|
CTD format Module 2 to 5: a/c to ICH CTD format. Module 1: Administrative & Product Information
Characteristics (SmPC),
|
CTD format. e-CTD recommended. Module 2 to 5: a/c to ICH CTD format. Module 1: regional requirements:
Characteristics (SmPC),
Certificate of pharmaceutical product (COPP). |
|
Document Required |
GMP Certificate |
GMP Certificate |
|
COPP/ Free Sale Certificate |
CPP or Free-Sales |
|
|
Application form |
Certificate of Analysis – Drug Substance/ Finished Product |
|
|
Site Master File |
Certificate of Analysis – Excipients |
|
|
BMR |
Alcohol - content declaration |
|
|
PSUR |
Pork – content declaration |
|
|
CoA (including Substance, Product) |
Certificate of Suitability for TSE |
|
|
- |
The diluents and colouring agents in the product formula |
|
|
- |
Patent Information |
|
|
- |
Letter of access or acknowledgement to DMF |
CONCLUSION:
Navigating the drug registration process in Kenya and the Saudi Arabia requires carefully attention to detail and adherence to regulatory requirements. Pharmaceutical companies must ensure that they compile all necessary documentation and data to support the safety, efficacy, and quality of their drugs. Additionally, companies should be prepared to cooperate with the regulatory authorities and undergo inspections of their manufacturing facilities to demonstrate the compliance with applicable standards. Overall, while the drug registration processes in Kenya and Saudi Arabia may present challenges, they are essential for safeguarding public health and ensuring that only safe and effective medications are available to patients in these countries. By understanding and effectively navigating these processes, pharmaceutical companies can successfully bring their products to market and contribute to improving healthcare outcomes in the region.
REFERENCES



Prakruti Pate*, Rahul Nayak, Dr. Maitreyi Zaveri, Dr. Vinit Movaliya, Drug Registration Requirement Process for Kenya and Saudi Arabia, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 4, 639-661 https://doi.org/10.5281/zenodo.15152956
 10.5281/zenodo.15152956
10.5281/zenodo.15152956
