Department of MBA (Pharmaceutical Management), D. Y. Patil Agriculture & Technical University, Talsande, Kolhapur
Artificial intelligence has emerged as a transformative force in pharmaceutical research, offering new opportunities to overcome long-standing challenges associated with cost, duration, and inefficiency in early drug discovery. Traditional discovery workflows often rely on labor-intensive experimentation and exhibit high attrition rates due to the limited predictability of biological targets and chemical candidates. Recent advances in machine learning, deep learning, generative modelling, and large language models have enabled a shift toward more predictive and data-driven strategies. These technologies facilitate comprehensive analysis of multi-omics datasets, accurate prediction of protein structures and ligand interactions, rapid identification of hit molecules, and rational design of leads with optimized pharmacological profiles. This review provides an in-depth examination of how artificial intelligence supports each stage of the drug discovery pipeline, from target identification and validation to hit generation and lead optimization. Emphasis is placed on the capabilities of modern computational frameworks, representative case studies, and the integration of AI-driven tools with experimental platforms. The discussion also highlights existing limitations, regulatory considerations, and future directions that will shape the adoption of AI in therapeutic development. Overall, the review underscores the growing potential of AI to accelerate innovation and improve decision-making across the drug discovery continuum.
Drug discovery is a complex and resource-intensive process that traditionally spans more than a decade from initial concept to regulatory approval. The early stages of discovery, which involve identifying biological targets, validating their therapeutic relevance, and generating lead molecules, are particularly prone to uncertainties and financial risk.[1] High attrition rates at these stages reflect the difficulty of predicting whether a target is truly disease-modifying or whether a chemical scaffold can be optimized into a safe and effective therapeutic candidate. Even with the introduction of high-throughput screening, combinatorial chemistry, and advanced structural biology, the discovery pipeline continues to face challenges related to data fragmentation, limited predictive accuracy, and inefficient exploration of the vast chemical space.[2]
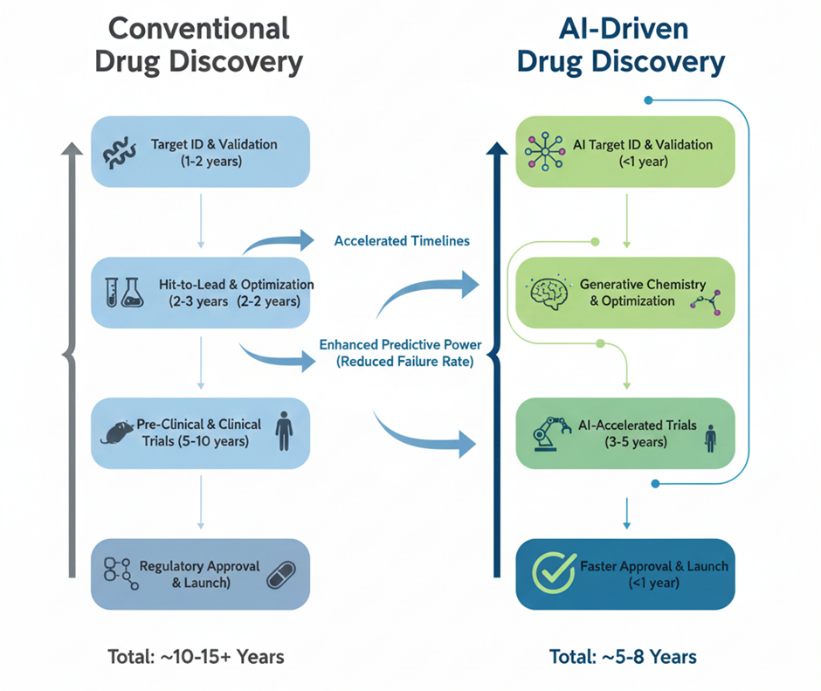
Fig 1. Schematic comparison of conventional drug discovery versus AI-driven approaches highlighting accelerated timelines and enhanced predictive power
Artificial intelligence has begun to transform this landscape by offering computational strategies capable of analyzing complex datasets, learning hidden relationships, and generating predictions with speed and precision. Unlike traditional computational methods, AI systems learn directly from data, allowing them to identify patterns that may be invisible to human experts or conventional statistical tools.[3] Machine learning and deep learning, in particular, have demonstrated remarkable success in predicting protein structures, analyzing multi-omics interactions, modelling ligand–protein binding, and guiding early optimization of drug-like properties. These methods reduce reliance on trial-and-error experimentation and enable researchers to prioritize the most promising biological targets and molecular candidates at an early stage.[4] The rapid evolution of generative AI has further accelerated the discovery process. Modern generative models can design entirely new chemical structures with predefined biological or pharmacokinetic characteristics, significantly reducing the time required to transition from hit identification to lead optimization. Similarly, reinforcement learning systems can iteratively refine molecular structures based on predefined objectives such as potency, selectivity, or synthetic feasibility.[5] Large language models trained on extensive biomedical and chemical corpora are now capable of assisting with retrosynthesis planning, literature summarization, and hypothesis generation, effectively functioning as intelligent research companions. AI is not merely a computational enhancement but represents a conceptual shift in the drug discovery paradigm. It enables more systematic, data-driven, and hypothesis-guided research in which biological interpretation and computational prediction reinforce each other.[6] The convergence of AI with high-content experimental platforms, automated laboratories, and rich biological datasets has created an opportunity for more predictive, efficient, and cost-effective discovery. As pharmaceutical research continues to generate vast amounts of heterogeneous data from genomic sequences to real-world clinical evidence AI becomes indispensable for integrating, interpreting, and translating this information into actionable insights.[7]
This review examines the role of AI across the entire drug discovery pipeline, beginning with target identification and progressing through hit discovery, lead generation, and lead optimization. Each stage is discussed with attention to emerging computational approaches, established tools, case examples, and the practical challenges that accompany their implementation. By synthesizing current advances and outlining future opportunities, the review highlights the growing impact of AI on therapeutic innovation and the potential of AI-driven strategies to reshape the future of pharmaceutical development.
2. OVERVIEW OF AI TECHNOLOGIES IN DRUG DISCOVERY
Artificial intelligence encompasses a broad spectrum of computational methodologies that enable automated pattern recognition, predictive modelling, and informed decision-making across complex and multidimensional datasets. In drug discovery, these technologies provide a framework for integrating chemical, biological, structural, and clinical data, thereby supporting rational design and early-stage prioritization of therapeutic candidates. Over the past decade, several categories of AI approaches have gained prominence within pharmaceutical research, each offering unique advantages depending on the nature of the data and the scientific question addressed.[8]
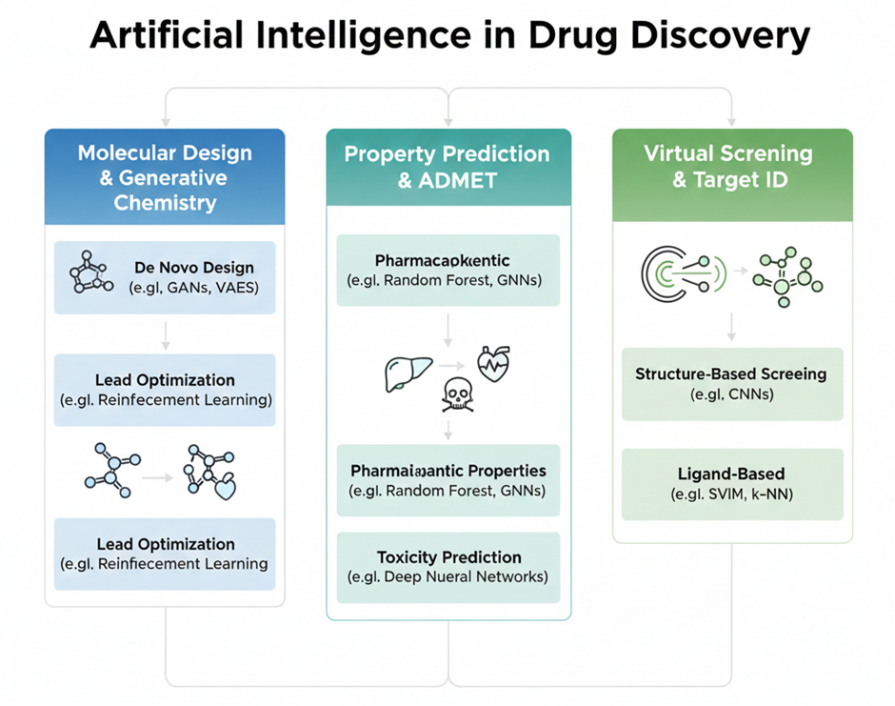
Fig 2. Classification of AI approaches used in drug discovery with typical use cases in molecular design, property prediction, and virtual screening.
Machine learning remains one of the foundational pillars of AI-driven drug discovery. Classical algorithms, such as random forests, support vector machines, logistic regression, and gradient-boosting frameworks, have demonstrated consistent performance across tasks involving classification, regression, and clustering. These methods are widely used to develop quantitative structure–activity relationship models, predict ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties, and refine virtual screening outputs. Their interpretable nature allows researchers to identify molecular descriptors and physicochemical attributes that influence biological activity, which enhances mechanistic understanding and supports decision-making in the early design stages.[9]
Table 1. Overview of Major AI Technologies Used in Drug Discovery
|
AI Technology |
Key Algorithms/ Models |
Primary Applications in Drug Discovery |
Strengths |
Limitations |
|
Machine Learning (ML) |
Random Forest, SVM, Gradient Boosting |
QSAR modeling, ADMET prediction, toxicity screening |
Interpretable, fast training, handles tabular data well |
Limited performance on high-dimensional data |
|
Deep Learning (DL) |
CNNs, RNNs, Transformers |
Structure prediction, bioactivity prediction, property modeling |
Strong pattern recognition, scalable to big data |
Requires large datasets; limited interpretability |
|
Generative Models |
GANs, VAEs, Diffusion Models, LLM-based chemical generators |
De novo molecule design, scaffold hopping, multi-parameter optimization |
Creates novel molecules, accelerates hit discovery |
Risk of unrealistic compounds; evaluation is challenging |
|
Reinforcement Learning (RL) |
DQN, PPO, Policy Gradient Methods |
Goal-driven molecule optimization, synthesis planning |
Supports iterative improvement toward desired properties |
Needs well-defined reward functions |
|
Large Language Models (LLMs) |
GPT-based, ChemBERTa, MolT5 |
Sequence–structure prediction, synthesis route generation, annotation |
Learns from diverse multimodal data, strong generalization |
Hallucination risks; requires curation |
|
Graph Neural Networks (GNNs) |
GCN, GAT, MPNN |
Molecular property prediction, docking score estimation |
Captures chemical graph structures effectively |
Limited by graph size, costly training |
|
Hybrid AI–Physics Models |
ML + Molecular Dynamics, ML + QM |
Binding affinity estimation, free-energy prediction |
Improves accuracy by integrating physics |
Slow and computationally intensive |
Deep learning has further advanced the capabilities of AI by capturing complex, nonlinear relationships that cannot be modelled effectively using traditional techniques. Convolutional neural networks and recurrent neural networks have been applied to image-based screening data, protein sequences, and molecular fingerprints. More recently, graph neural networks have emerged as one of the most powerful classes of models in medicinal chemistry. These networks treat molecules as graphs of atoms and bonds, enabling them to learn directly from structural topology rather than relying on predefined descriptors. Their ability to accurately predict protein–ligand binding affinities, physicochemical properties, and toxicity profiles has made them indispensable tools for high-throughput computational screening and lead prioritization.[10]
Generative artificial intelligence represents another transformative development. Using architectures such as variational autoencoders, generative adversarial networks, and diffusion models, generative AI systems can propose entirely new chemical structures that satisfy predefined constraints related to potency, selectivity, or drug-likeness. These models explore chemical space far more efficiently than traditional enumeration methods and generate novel scaffolds that may not exist in current medicinal chemistry libraries. In parallel, reinforcement learning techniques guide iterative molecular optimization by rewarding models for producing compounds with desirable attributes. This has enabled automated molecular refinement and the creation of multi-objective optimization frameworks capable of balancing potency, toxicity, solubility, and synthetic feasibility.[11] Large language models have recently emerged as versatile tools that support several stages of the drug discovery process. Trained on extensive chemical, biological, and biomedical literature, these models can interpret protein mutations, generate structural hypotheses, assist with retrosynthesis planning, summarize research findings, and enhance knowledge extraction from unstructured datasets. Their ability to integrate textual, sequence, and molecular data provides a foundation for multimodal reasoning and accelerates hypothesis generation in the early discovery phase.[12] Together, these AI technologies create an integrated computational environment that enhances predictive accuracy, improves efficiency, and reduces uncertainty during drug discovery. By complementing experimental approaches with scalable and data-driven analysis, AI serves as a catalyst for innovation and enables researchers to navigate complex biological and chemical landscapes with greater confidence.[13]
3. AI IN TARGET IDENTIFICATION
Target identification represents a critical first step in the drug discovery pipeline, as it sets the direction for downstream research and determines the overall likelihood of therapeutic success. Conventional approaches rely heavily on experimental screening, literature-based evidence, and expert interpretation of biological pathways. Although these methods have contributed significantly to modern therapeutics, they often struggle to interpret large-scale datasets, uncover hidden biological relationships, or detect subtle regulatory elements that drive disease progression. Artificial intelligence has emerged as a powerful solution capable of integrating heterogeneous datasets, extracting meaningful biological signatures, and prioritizing targets with a higher probability of clinical relevance.[14] One of the most significant contributions of AI in this domain is the analysis of multi-omics datasets. Advances in next-generation sequencing and high-throughput analytical platforms have generated vast amounts of genomic, transcriptomic, proteomic, and metabolomic data. Traditional statistical models are often insufficient for capturing the complex, nonlinear interactions within these datasets. Machine learning algorithms, in contrast, can identify differential expression patterns, mutation clusters, epigenetic alterations, and disease-specific biomarkers with remarkable accuracy. Deep learning further enhances this capability by discovering latent biological features that may be overlooked by conventional methods. These computational insights enable researchers to shortlist genes or proteins that play central roles in disease mechanisms.[15]
Network biology has also benefited from AI-driven advancements. Biological systems operate through intricate networks of interacting molecules rather than isolated pathways. AI-based network analysis tools examine protein–protein interactions, signal transduction cascades, transcriptional regulation, and metabolic pathways to highlight nodes with high connectivity or regulatory influence. Graph neural networks, in particular, have shown strong performance in modeling network topologies and predicting the functional importance of specific nodes. These models help identify targets that are essential for disease survival or progression and may reveal intervention points that conventional pathway analysis fails to detect.[16] AI also supports target identification through large-scale literature mining and natural language processing. Scientific literature, clinical trial data, and electronic health records contain rich insights but are often too voluminous for manual review. Modern language models trained on biomedical corpora can extract disease–gene associations, interpret mechanistic hypotheses, highlight contradictory findings, and map emerging therapeutic trends. This automated knowledge extraction accelerates the discovery of novel targets and supports evidence-driven prioritization.[17] Structural insights generated by AI have strengthened the reliability of target identification. Protein structure prediction tools such as AlphaFold2 and RoseTTAFold provide high-resolution structural models that assist researchers in assessing druggability, identifying potential binding pockets, and evaluating conformational dynamics. These structural predictions enhance biological interpretation and offer early indicators of whether a target is likely to support small-molecule or biologic intervention.[18] Case studies across therapeutic areas underscore the practical value of AI in target identification. AI-based platforms used by companies such as BenevolentAI, Exscientia, and Insilico Medicine have successfully uncovered targets in oncology, neurodegeneration, and inflammatory diseases that were either previously unknown or not fully appreciated. These real-world examples highlight the capacity of AI to accelerate hypothesis generation and provide a systematic foundation for initiating drug discovery programs. Overall, AI enhances target identification by integrating diverse data sources, revealing novel biological relationships, and enabling more informed decision-making. Its ability to reduce uncertainty, expand the search space, and uncover hidden patterns positions AI as an indispensable tool for initiating modern drug discovery efforts.[19]
4. AI IN TARGET VALIDATION
Target validation is essential for confirming that a proposed biological target plays a causal or regulatory role in disease progression and can be modulated safely and effectively. While traditional validation approaches rely on genetic knockdown, animal models, biochemical assays, and phenotypic screening, these methods are time-consuming, expensive, and often limited by biological complexity. Artificial intelligence has reshaped this phase of drug discovery by improving predictive precision, strengthening mechanistic understanding, and supporting evidence-based decision-making before significant resources are invested in downstream development.[20] One of the most transformative AI contributions to target validation lies in structural biology. Tools such as AlphaFold2 and RoseTTAFold now provide highly accurate three-dimensional protein structures for targets that lack experimental crystallographic or cryo-EM data. Accurate structural information is indispensable for assessing ligand-binding sites, evaluating target druggability, predicting allosteric regions, and understanding conformational flexibility. AI-generated structures greatly reduce the reliance on slow and technically demanding laboratory procedures, allowing researchers to rapidly evaluate whether a target is likely to support small-molecule or biologic intervention. These models also facilitate early docking simulations, molecular dynamics studies, and binding site prioritization, further strengthening the biological rationale for pursuing a target.[21]
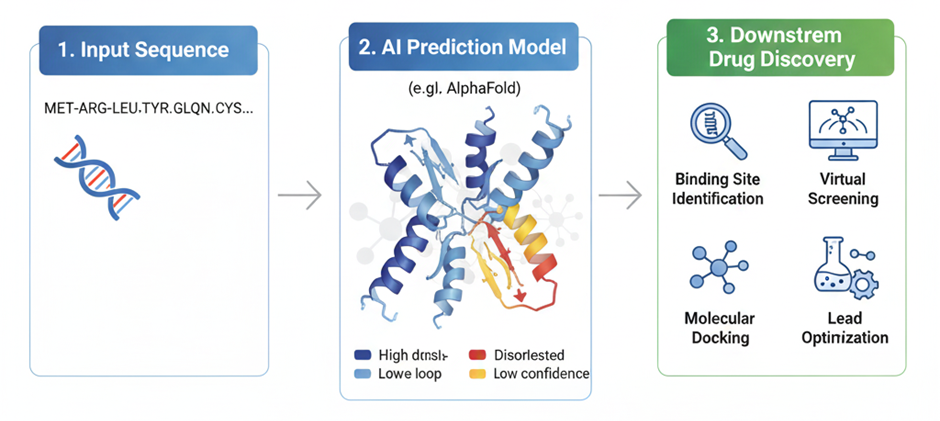
Fig 3. AI-Driven Protein Structure Prediction and Target Validation.
AI also enhances target validation through predictive modelling of functional relevance. Machine learning algorithms integrate genetic, clinical, and phenotypic datasets to determine whether perturbing a target is likely to produce a desirable therapeutic effect. For example, ML models trained on CRISPR screening data, patient-derived gene expression profiles, and multi-omic biomarkers can identify essential genes, synthetic lethality relationships, and compensatory pathways that may influence therapeutic outcomes. These computational assessments help distinguish between primary disease drivers and secondary responders, thereby guiding researchers toward intervention points with the highest clinical potential.[22] In addition, AI plays a key role in predicting off-target activity and safety risks associated with target modulation. Deep learning models trained on toxicogenomic, pharmacovigilance, and phenotypic assay data can forecast potential liabilities such as cardiotoxicity, hepatotoxicity, immune activation, or unintended pathway interference. Such early safety predictions reduce the risk of costly attrition in later stages and provide insight into whether a target is likely to exhibit acceptable pharmacological and toxicological behavior. By integrating adverse event databases and real-world clinical data, AI offers a more comprehensive risk evaluation than traditional laboratory methods alone.[23]
Natural language processing provides another layer of validation by synthesizing evidence dispersed across biomedical literature, patents, clinical trials, and electronic health records. AI-driven text mining can detect inconsistent findings, identify understudied target functions, highlight gaps in mechanistic knowledge, and consolidate confidence levels based on existing evidence. This automated aggregation of global knowledge supports a more objective and data-rich evaluation of target credibility.[24] Practical examples highlight the growing influence of AI-based validation strategies. Several biotechnology companies have successfully used AI platforms to validate targets in oncology, neurodegenerative disorders, fibrosis, and autoimmune diseases. For instance, AI-guided analysis of multi-omic and patient-level data has helped identify pathogenic signaling nodes in cancer subtypes and validate targets for drug repurposing in inflammatory conditions. These achievements illustrate how AI reduces uncertainty by grounding target selection in computational rigor and real-world biological evidence. In summary, AI significantly strengthens target validation by integrating structural, functional, clinical, and safety perspectives. Its predictive capabilities, combined with its ability to incorporate diverse datasets, allow researchers to prioritize targets with higher therapeutic relevance and lower risk profiles. This leads to more informed decision-making, fewer failures in later development stages, and a more efficient transition from early discovery to hit identification.[25]
Table 2. Examples of AI Platforms in Pharmaceutical Applications
|
Platform / Tool |
Developer |
AI Method |
Application Area |
Notable Achievements |
|
AlphaFold2 |
DeepMind |
Transformer-based DL |
Protein structure prediction |
>90% accuracy in CASP14 |
|
RosettaFold |
University of Washington |
DL + Structure-aware modeling |
Protein folding |
Efficient alternative to AlphaFold |
|
Exscientia Platform |
Exscientia |
ML + Generative AI |
Lead discovery & optimization |
First AI-designed drug entered clinical trials |
|
Atomwise AtomNet |
Atomwise |
CNN-based DL |
Structure-based virtual screening |
Screened >16 billion compounds |
|
Insilico Medicine’s Chemistry42 |
Insilico Medicine |
GAN + RL |
De novo molecule generation |
Produced multiple preclinical candidates |
|
Schrödinger AI Suite |
Schrödinger |
ML-integrated simulations |
Property prediction, binding affinity |
Improved free-energy calculations |
|
IBM RXN for Chemistry |
IBM |
Neural machine translation |
Synthesis prediction |
Produces human-level retrosynthesis plans |
5. AI IN HIT DISCOVERY AND VIRTUAL SCREENING
Artificial intelligence has substantially reshaped the landscape of hit discovery by introducing computational models capable of navigating vast chemical spaces with unprecedented efficiency and accuracy. In conventional workflows, hit identification involves high-throughput screening (HTS) of large compound libraries, an approach that is both resource-intensive and limited by the physical number of molecules available for testing. AI-driven methods particularly machine learning (ML) and deep learning (DL) architectures enable rapid digital screening of millions to billions of compounds, significantly reducing experimental load while enriching high-quality hit candidates. These models excel in pattern recognition, enabling them to predict activity, filter drug-like molecules, and identify novel chemotypes that may be overlooked by traditional similarity-based methods.[26] AI-augmented virtual screening can be broadly classified into ligand-based approaches and structure-based approaches, each benefiting from advanced computational innovations. Ligand-based virtual screening uses quantitative structure–activity relationship (QSAR) models, deep neural networks, and graph-based learning to derive predictive rules from known active molecules. Such models can rapidly evaluate untested compounds for probability of binding or biological activity, even in cases where structural data for the target protein remain limited. On the other hand, structure-based screening has evolved through integration with AI-driven protein modelling techniques, including advanced algorithms capable of predicting three-dimensional protein structures with near-experimental accuracy. These tools facilitate more precise docking, scoring, and binding affinity prediction, thereby enhancing the reliability of hit identification.[27]
Generative AI plays an increasingly central role in expanding the scope of hit discovery. Models such as variational autoencoders, generative adversarial networks, and transformer-based architectures can design novel molecular structures optimized for target interaction, physicochemical properties, and synthetic accessibility. By learning from large chemical and bioactivity datasets, these generative systems produce virtual compounds that diverge from existing libraries while exhibiting biologically meaningful features. This approach not only accelerates hit discovery but also supports exploration of new chemical space outside traditional medicinal chemistry boundaries.[28] Despite its promise, AI-driven hit identification faces challenges, including biases in training datasets, inaccuracies in predicted binding affinities, and the need for experimental validation. Hybrid workflows that combine AI predictions with high-throughput experimentation, microfluidics, and fragment-based screening are becoming increasingly common to mitigate these limitations. As AI models continue to evolve through incorporation of richer bioactivity data, improved scoring functions, and multimodal learning frameworks, their role in accelerating early-phase drug discovery will continue to strengthen. Ultimately, AI-enabled virtual screening fosters a more efficient, cost-effective, and exploratory approach to identifying structurally diverse and pharmacologically relevant hits.[29]
6. AI IN LEAD OPTIMIZATION
Lead optimization represents one of the most resource-intensive stages of drug discovery, requiring iterative refinement of molecular hits to enhance potency, selectivity, pharmacokinetic behaviour, and safety. Artificial intelligence has significantly transformed this process by offering predictive, generative, and optimization frameworks that guide medicinal chemists in designing superior lead candidates with higher efficiency and precision. Instead of relying exclusively on manual structural modifications and experimental cycles, AI models can learn from historical datasets, anticipate molecular liabilities, and propose modifications that align with desired therapeutic profiles. Machine learning–based property prediction models are central to AI-enabled lead optimization.[30] These include regression and classification algorithms capable of estimating key ADMET (absorption, distribution, metabolism, excretion, toxicity) parameters, binding affinities, solubility, permeability, and metabolic stability. Deep learning architectures especially graph neural networks (GNNs), message-passing neural networks (MPNNs), and transformer-based molecular encoders offer enhanced accuracy by capturing structural complexity at the atom and bond level.[34] By enabling early identification of liabilities such as cardiotoxicity, CYP450 interactions, or poor oral bioavailability, AI models help prioritize only those candidates likely to succeed downstream. The integration of AI with structure-based design further strengthens optimization by offering insights into ligand–protein interactions at atomic resolution. Deep learning–enhanced docking, molecular dynamics simulation, and binding free energy prediction tools facilitate rapid evaluation of structural modifications and help chemists identify the most favourable interaction patterns. These computational insights reduce the number of physical synthesis cycles needed to validate hypotheses, accelerating convergence toward optimal leads. A major advancement in recent years is the rise of AI-driven generative chemistry, where models not only evaluate molecules but also propose new ones.[31] Variational autoencoders, generative adversarial networks, diffusion models, and reinforcement learning frameworks can generate molecules optimized simultaneously for activity, physicochemical properties, and synthetic feasibility. Reinforcement learning in particular enables goal-directed optimization by rewarding models for producing structures that meet multiple predefined criteria. When combined with retrosynthetic planning tools, AI systems can also suggest viable synthetic routes for proposed molecules, making them more practical for laboratory synthesis. Despite substantial progress, challenges remain in ensuring that AI-designed leads translate effectively in vivo. Data quality, limited representation of rare chemical motifs, lack of experimental feedback loops, and model interpretability continue to affect prediction accuracy. Hybrid workflows that integrate AI predictions with high-throughput synthesis, autonomous experimentation platforms, and iterative learning cycles are emerging as effective solutions to bridge this gap. These closed-loop systems allow AI models to continuously learn from newly generated experimental data, improving their performance over time.[32]
7. CASE STUDIES DEMONSTRATING THE IMPACT OF AI IN DRUG DISCOVERY
The practical value of artificial intelligence in drug discovery is best illustrated through real-world case studies that demonstrate tangible acceleration of timelines, identification of novel chemical entities, and successful transition of AI-generated candidates into preclinical and clinical development. Over the past decade, collaborations between pharmaceutical companies, biotech start-ups, and AI-focused firms have produced several breakthroughs that validate the transformative potential of machine learning and deep learning in early-stage research.[33] These case studies highlight diverse applications from target identification to de novo molecule generation and collectively show how AI can bridge scientific gaps that historically constrained traditional methods.[34] One of the most prominent examples is the development of DSP-1181 by Exscientia in collaboration with Sumitomo Dainippon Pharma. DSP-1181, a selective serotonin 5-HT1A receptor agonist for obsessive-compulsive disorder, became the first AI-designed molecule to enter human clinical trials. By leveraging deep learning–driven design cycles and multi-objective optimization, the research team reduced the lead optimization timeline from an estimated 4–5 years to less than 12 months. This achievement demonstrated how AI can evaluate millions of design options, prioritize molecules with optimal pharmacokinetic and safety profiles, and streamline experimental validation workflows.[35] Another notable case involves In-silico Medicine, which used generative adversarial networks and reinforcement learning to develop a novel inhibitor of discoidin domain receptor 1 (DDR1), a target implicated in fibrosis. Starting from target identification, In-silico used its integrated AI platform to generate, optimize, and score candidate molecules. Within 46 days, the team identified potent lead compounds and synthesized several promising candidates a process that traditionally requires several months. Subsequent in vivo studies confirmed strong anti-fibrotic activity, demonstrating the predictive strength of AI-generated molecular designs.[36] In the domain of protein structure prediction, the introduction of AlphaFold2 by DeepMind represents a landmark milestone. AlphaFold2’s ability to accurately predict protein three-dimensional structures has accelerated target understanding, binding site mapping, and rational design processes. Numerous pharmaceutical groups have incorporated AlphaFold2 predictions into early discovery workflows, particularly for proteins lacking experimental structures, thereby enabling structure-based drug design at a scale and precision previously unattainable. AI has also demonstrated value in repurposing existing drugs, especially during time-sensitive global health challenges.[37] During the COVID-19 pandemic, machine learning platforms were rapidly deployed to screen antiviral compound libraries, prioritize host-target interactions, and identify repurposing candidates. BenevolentAI notably used its knowledge graph and predictive modelling pipeline to identify baricitinib as a potential therapeutic agent for SARS-CoV-2 infection. The drug later progressed into clinical trials and obtained emergency authorization, illustrating how AI can accelerate therapeutic responses during emerging public health crises.[38]
Collectively, these case studies highlight the practical strengths of AI: speed, predictive accuracy, and the ability to explore chemical and biological space beyond human intuition. While not all AI-generated candidates advance successfully through clinical development, the reductions in cycle times, improved hit-to-lead efficiency, and generation of innovative chemical structures firmly establish AI as a critical enabler of modern pharmaceutical research. As computational models continue to improve and integrate with robotic automation, omics technologies, and high-throughput experimentation, the number and quality of AI-driven success stories are expected to grow substantially.[39]
8. CHALLENGES, LIMITATIONS, AND REGULATORY CONSIDERATIONS IN AI-DRIVEN DRUG DISCOVERY
Despite its rapid advancement and growing adoption, AI-driven drug discovery faces several scientific, technical, ethical, and regulatory challenges that collectively shape its progress and real-world impact. While AI has demonstrated remarkable potential to accelerate early discovery and improve decision-making, its deployment in pharmaceutical research is constrained by limitations associated with data quality, model interpretability, reproducibility, and governance. Understanding these challenges is essential for developing robust frameworks that ensure AI-derived insights remain reliable, transparent, and scientifically valid A critical limitation lies in the quality and availability of training datasets. AI models rely heavily on large, diverse, and structurally rich datasets to learn meaningful patterns. However, many bioactivities, structural, and ADMET datasets contain inconsistencies, experimental variability, measurement noise, or incomplete annotations. In addition, negative data (inactive or toxic molecules) are far less frequently reported, creating imbalanced datasets that can skew model performance. Proprietary pharmaceutical datasets are often inaccessible, further limiting opportunities to train models capable of generalizing across targets and therapeutic areas. As a result, predictions may perform well on internal benchmarks but fail when exposed to novel chemical structures or biological systems Interpretability and transparency represent another major concern. Many high-performing models, particularly deep learning architectures such as graph neural networks and transformers, operate as "black boxes" with limited insight into the reasoning behind their predictions. For medicinal chemists and regulatory agencies, the inability to explain why a model proposes a certain molecule or predicts a specific property poses a barrier to trust and adoption. Efforts to integrate explainable AI (XAI) approaches are growing, but current methods often remain insufficient for capturing the complexity of molecular representations and biological networks. Furthermore, generalizability and reproducibility continue to present challenges. AI models trained on narrow chemical or biological domains may perform poorly when applied to new targets or novel chemotypes. Reproducibility is hindered by differences in data preprocessing, parameter settings, evaluation metrics, and benchmark datasets across research groups. Standardized workflows and reporting guidelines are urgently needed to ensure that models can be independently validated and compared across platforms. On the regulatory front, the emergence of AI-designed molecules introduces new considerations for drug approval pathways. Traditional regulatory frameworks were developed for molecules designed through conventional experimentation and may not fully address issues related to algorithm-driven decision-making. Agencies such as the FDA and EMA have begun introducing guidance surrounding AI in medical devices, yet formal policies for AI-driven drug design remain in their early stages. Key questions include accountability for model predictions, documentation requirements for training data, validation standards for computationally generated molecules, and oversight mechanisms to evaluate the robustness of AI-assisted decisions. The establishment of regulatory sandboxes and adaptive frameworks is anticipated to facilitate clearer pathways for AI-enabled drug candidates.[40]
9. FUTURE PROSPECTS OF AI IN DRUG DISCOVERY
The application of artificial intelligence in drug discovery is poised to undergo significant evolution, offering transformative potential across the entire therapeutic development pipeline. As computational capabilities, algorithmic sophistication, and data availability continue to expand, AI is expected not only to accelerate traditional workflows but also to enable novel paradigms that redefine how drugs are discovered, designed, and developed. One of the most promising future directions is the integration of multimodal data sources. Current AI applications often rely on either chemical, biological, or clinical data streams independently. However, the convergence of genomics, transcriptomics, proteomics, metabolomics, imaging data, and real-world patient records can provide a more holistic understanding of disease mechanisms and therapeutic responses. Advanced AI models capable of handling multimodal data will allow simultaneous learning across these heterogeneous datasets, enabling precise target identification, personalized therapy design, and adaptive optimization of leads. Generative and self-learning AI systems are also expected to expand the frontiers of molecular innovation.[41]
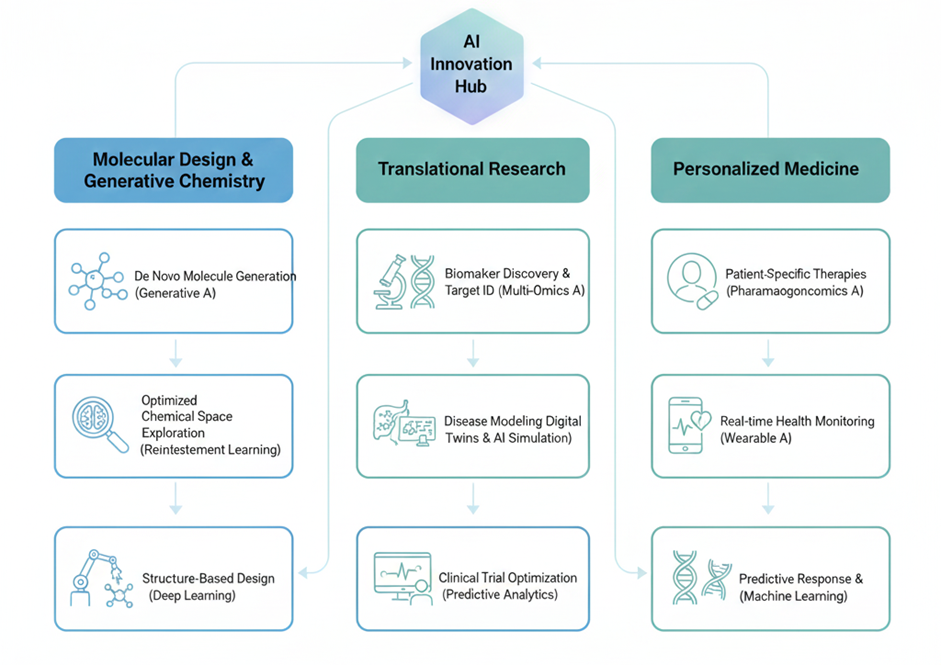
Fig 4. Future trends and opportunities in AI-driven drug discovery
Next-generation generative models, coupled with reinforcement learning and closed-loop experimental validation, will facilitate autonomous drug design that iteratively refines chemical structures based on experimental feedback. These systems have the potential to explore unprecedented regions of chemical space, generate structurally novel scaffolds, and optimize multi-objective profiles including potency, selectivity, pharmacokinetics, and safety far more efficiently than conventional medicinal chemistry approaches.
10. CONCLUSION
Artificial intelligence has emerged as a transformative force in modern drug discovery, fundamentally reshaping the strategies for target identification, validation, hit discovery, and lead optimization. By leveraging machine learning, deep learning, generative models, and large language models, researchers can navigate complex biological and chemical landscapes with unprecedented speed, accuracy, and efficiency. AI-driven approaches enable the integration of heterogeneous datasets, identification of novel therapeutic targets, prediction of molecular properties, and rational design of optimized lead compounds, thereby reducing the reliance on traditional trial-and-error methods and minimizing attrition rates. The practical impact of AI is evident through numerous case studies that demonstrate accelerated timelines, successful generation of novel chemical entities, and improved translational outcomes. However, significant challenges remain, including data quality limitations, model interpretability, reproducibility, and the need for regulatory clarity. Addressing these challenges through robust data curation, explainable AI techniques, hybrid computational-experimental workflows, and collaborative regulatory frameworks is essential to fully harness AI’s potential. Looking forward, the convergence of AI with multimodal datasets, autonomous laboratories, and precision medicine platforms promises to redefine the drug discovery paradigm. As computational models continue to evolve, AI is poised not only to enhance efficiency but also to enable innovative therapeutic designs that were previously unattainable. In this context, AI represents both a catalyst and a partner in the pursuit of safer, more effective, and personalized medicines, underscoring its critical role in the future of pharmaceutical research and development.
REFERENCES


Avdhut Manjare, Akash Malakane, Shravani Girigosavi, AI-Driven Drug Discovery: From Target Identification to Lead Optimization, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 3, 3203-3218. https://doi.org/10.5281/zenodo.19229043
 10.5281/zenodo.19229043
10.5281/zenodo.19229043
