1,2,3,4Department of Bachelor of Pharmacy, Kasturi Shikshan Sanstha’s College of Pharmacy, Shikrapur, Pune
5Department of Master of Pharmacy, Kasturi Shikshan Sanstha’s College of Pharmacy, Shikrapur, Pune
Effective information management is inextricably tied to pharmacovigilance, which is the science and practice of identifying, evaluating, comprehending, and preventing side effects or other drug-related issues. Information plays a crucial part in pharmacovigilance, which includes gathering, analyzing, and disseminating data for the best possible patient safety. Pharmacovigilance is based on a strong information system that makes it easier to gather reports of adverse events from patients, healthcare providers, and other stakeholders. the development of data sources, with a focus on combining wearable technology, electronic health records, and empirical data to increase the volume and variety of data that can be analyzed. By automating signal identification and predictive modeling, cutting-edge technologies like artificial intelligence and machine learning are revolutionizing pharmacovigilance. How to use those technologies to sort through large datasets, find any safety issues, and support regulatory decision-making. The abstract also explores the significance of organized information exchange among pharmaceutical corporations, regulatory bodies, and healthcare providers. Effective risk management strategies can be developed and a proactive reaction to new risks is ensured by timely and transparent safety information transmission.
Pharmacovigilance is a critical component of clinical research, aimed at ensuring the safety and efficacy of medicinal products throughout their lifecycle. It involves the detection, assessment, understanding, and prevention of adverse drug
reactions (ADRs) and other drug-related problems. This process is essential for identifying potential risks associated with pharmaceutical interventions, ensuring regulatory compliance, and safeguarding public health. In clinical trials, pharmacovigilance focuses on collecting and analyzing safety data to detect early signals of adverse effects. These activities include monitoring suspected ADRs, reporting safety findings to regulatory authorities, and developing risk management strategies. Advances in technology, such as data mining and artificial intelligence, have improved the efficiency and accuracy of signal detection and risk assessment.The integration of pharmacovigilance into clinical research strengthens post-market surveillance, enhances patient outcomes, and informs healthcare decision-making. As the global pharmaceutical landscape evolves, continuous efforts are needed to improve pharmacovigilance practices, address emerging challenges, and foster international collaboration to ensure the safe use of medicines. [1]
HISTORY:
1.Thalidomide Tragedy (1950s–1960s): Thousands of babies were born with serious birth problems as a result of the drug, which was first recommended as a sedative and antiemetic. Increased awareness of the possible harm that medicines could cause, particularly during pregnancy, resulted from the aftermath.
2. Creation of WHO Program (1968): In 1968, WHO created the international Drug Monitoring Program in reaction to the thalidomide disaster. By encouraging cooperation in gathering and evaluating data on adverse drug reactions (ADRs), this program established the framework for an international network of pharmacovigilance centres.
3. The Adverse Event Reporting System (AERS) was established by the FDA in the 1970s: The FDA can now monitor and control drug safety in the United States because to AERS, a crucial tool for gathering, organising, and evaluating data on adverse events linked to medications.
4. ICH Guidelines (1990s): Pharmacovigilance procedures were standardized worldwide in large part because to the International Conference on Harmonization (ICH). ICH guidelines, like E2B, promoted global collaboration among regulatory bodies by offering a standardized framework for the gathering and sharing of safety data.
5. Pharmacovigilance system (2005): To improve the oversight and monitoring of pharmaceuticals, the European Union implemented a thorough pharmacovigilance system. The European Medicines Agency (EMA) was instrumental in organizing risk management plans and safety evaluations.
6. Periodic safety update reports (PSURs): These reports are now required of all holders of marketing authorizations. By regularly submitting safety data to regulatory bodies, these reports guarantee ongoing assessment of a drug's safety profile over the course of its lifecycle.
OBJECTIVE:
1) Monitoring Adverse Effects: Maintains monitoring to identify and assess adverse drug responses (ADRs) that occur when taking prescription drugs.
2) Evaluation of Risk and Benefits: Examining a medication's risk-to-benefit ratio to make sure that the therapeutic advantages outweigh any possible hazards or negative effects.
3) Data Collection and Analysis: Methodically compiling and examining data on medication safety, such as patient, healthcare provider, and clinical trial reports.
4) Risk Management and Mitigation: Creating plans for risk reduction, changing labels, or, if required, removing medications from the market as a means of managing and reducing the hazards connected with them.
5) Encouraging Safe Medication Use: Teaching patients and medical professionals on proper dosage, administration, and monitoring for the safe and efficient use of pharmaceuticals.
6) Communication and Information
Dissemination: Encouraging the public, regulatory bodies, and healthcare providers to receive safety information so they can make educated decisions regarding the use of medications.
7) Regulatory Compliance: Making sure that the rules governing drug safety reporting and monitoring are followed. After a medication is licensed and put on the market, post-marketing surveillance keeps an eye on its safety to spot uncommon or long-term side effects that might not have been noticeable during clinical studies.
Constitute Pharmacovigilance Program of India [13]: The Pharmacovigilance Programme of India (PvPI) is an initiative by the Ministry of Health and Family Welfare, Government of India, aimed at ensuring drug safety by monitoring adverse drug reactions (ADRs) and other related aspects. It is managed by the Indian Pharmacopoeia Commission (IPC) under the aegis of the Central Drugs Standard Control Organization (CDSCO).The Pharmacovigilance Programme of India (PvPI) was launched in 2010 by the Ministry of Health and Family Welfare (MoHFW), Government of India. It is aimed at monitoring the safety of medicines and ensuring their safe use in the Indian population. The concept of pharmacovigilance in India was first initiated under the National Pharmacovigilance Programme (NPP) in 2004, but its scope was limited.PvPI was launched in July 2010 to expand pharmacovigilance activities and make them more robust.In 2011, PvPI was transferred from the All India Institute of Medical Sciences (AIIMS), New Delhi to the Indian Pharmacopoeia Commission (IPC), Ghaziabad, which now serves as the National Coordinating Centre (NCC).
Clinical Research History [16]: Clinical research is a broad strategy for increasing medical knowledge and enhancing patient outcomes. Preclinical research entails laboratory tests to investigate the safety and possible effectiveness of interventions in the early stages. In order to collect preliminary data, this step frequently involves testing on cell cultures, animal models, or computer simulations. Researchers painstakingly create a protocol outlining study objectives, inclusions/exclusion criteria, and reliable techniques if encouraging results are received. After participants are fully educated about the goals, possible hazards, and advantages of the study, their informed consent is obtained, upholding the ethical foundation of clinical research. A crucial step in making sure that the research complies with established norms and ethical standards is obtaining regulatory approval from ethics committees and other pertinent authorities. Phase I of clinical trials focuses on safety; Phase II involves extending the study to a larger cohort to further evaluate safety and efficacy; and Phase III involves large-scale trials that compare therapies to current standards of care (Figure10). In these stages, the randomized controlled trial (RCT) design is frequently used to randomly assign people to intervention or control groups, improving the dependability of results.
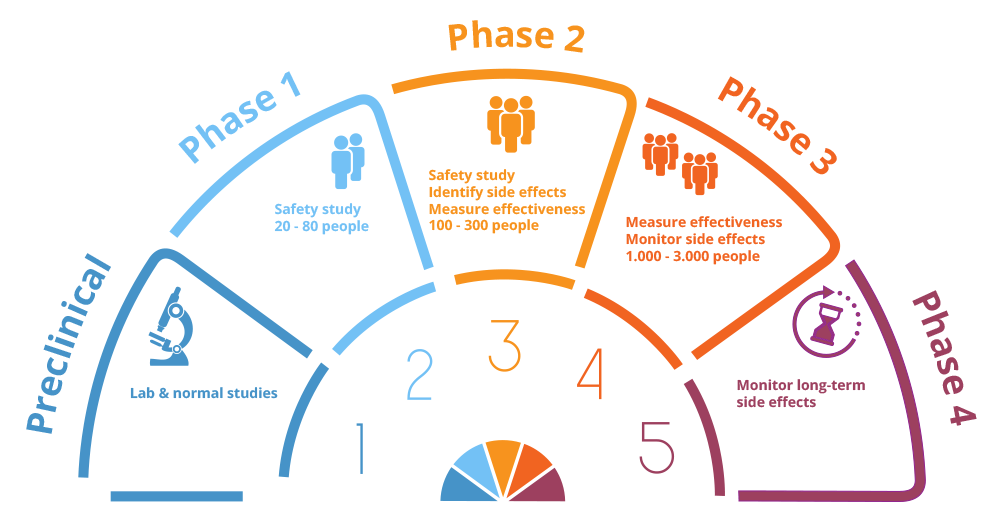
Fig. Phases of Clinical Trails
Phases 0f Clinical Trails [12,15]:
Phase:
Goals: To investigate human behaviour. Assess fundamental attributes such as ADME.Participants: Ten to fifteen healthy participants in very small groups Take a very small dose. Duration: Brief (usually a few days) End Points: A validated test that accurately measures a disease's progression must be in place before phase 0 studies that aim to measure that progression can begin (PD end point). Activities: Involve administering subtherapeutic doses by microdosing. Sample Size: Phase 0 trials are usually small-scale, with fewer than 15 individuals, and the medication is given for a short period oftime. Regulatory approval: Phase 0 trials are not a formal stage of development and do not need FDA permission. Their nature is exploratory.
Phase 1:
Goals: Establish safety, dosage range, and any adverse effects. comprehends pharmacokinetics and medication metabolism. Participants: persons with the target conditions or a small sample of 22–100 healthy volunteers. Dose: concentrating on small groups to evaluate the safety profile of the treatment. Time frame: a few months. End goal: Phase 1 studies seek to determine limiting toxicities, identify side effects, establish safe dosages, and use laboratory testing to identify organ-related harm.Activities: Give dosages in order to determine a range of safe dosages. Keep an eye on pharmacokinetics, pharmacodynamics, and side effects. Sample Size: Typically, a phase 1 trial's sample size included more than 20 participants. Regulatory Approval: Subject to regulatory agencies and requires regulatory approvals.
Phase 2:
Goals: Phase 2 trials start to gauge the drug's efficacy and further assess safety. Participants: People who have the ailment that the medication is meant to cure.Dose: Given at or close to the recommended therapeutic dosage. Final Goal: Assess the effectiveness and adverse effects. Keep an eye on safety.Activities: Evaluate the efficacy and safety of treatment.Sample Size: Usually includes fewer than fifty patients. Regulatory Approval: The endorsement of organizations such as the FDA or EMA.
Phase 3:
Goals: Phase 3 studies verify efficacy, monitor adverse effects, and contrast the novel treatment with accepted practices.Participants: A sizable patient population having the ailment the medication is meant to treat.Dosage: Like phase 2. Give the recommended therapeutic dose or very close to it. Phase 3 trials may last for a number of years. Final Goal: Evaluate overall advantages versus risk, efficacy, and mild side effects. Get more safety-related information. Activities: Compare to usual therapies, observe for side effects, and test treatment in large numbers to ensure efficacy. There are between 100 and 1000 people in the sample. Regulatory Approval: Phase 3 trials serve as important treatment evaluations. They examine side effects, efficacy, and safety.
Phase 4:
Goals: Phase 4 studies take place following a drug's approval and release onto the market. Participants: has the potential to affect millions of patients. Dosage: Reflects actual usage. Duration: Phase 4 trials, which track the medication in real-world situations, can go on indefinitely. Final Goal: Assess long-term efficacy and safety. may reveal long-term or rear impacts. Activities: Following approvals, phase 4 trials take place. They evaluate novel applications, safety, and effectiveness in a range of demographics. Because of the real world, the sample size may be very huge. Regulatory Approval: Note that these are only broad guidelines, and specifics may vary greatly based on the condition and therapy type under investigation.
Drugs Controiier General of India [3,7]: The regulatory body in India in charge of approving pharmaceuticals, including medications and medical equipment, is the Drug Controller General of India (DCGI). Its function in pharmacovigilance entails tracking and evaluating medications' safety after they are marketed. It looks into adverse drug responses that consumers and medical experts report, and it makes sure pharmaceutical corporations follow safety rules. The Central Drugs Standard Control Organization (CDSCO), which was established in 1940, is where DCGI's history begins. As CDSCO developed over time, the Drugs and Cosmetics Act of 1961 was passed, giving the agency the authority to control the import, manufacturing, distribution, and sale of pharmaceuticals.The Drugs Controller General of India (DCGI) is the head of the Central Drugs Standard Control Organization (CDSCO), which is part of the Ministry of Health and Family Welfare, Government of India. The DCGI plays a crucial role in ensuring the safety, efficacy, and quality of drugs, medical devices, and cosmetics in the country.
Functions:
1. Regulatory Approvals: Oversees the approval process for new drugs, vaccines, medical devices, and clinical trials in India. Ensures compliance with the Drugs and Cosmetics Act, 1940, and its rules.
2. Pharmacovigilance: Monitors adverse drug reactions (ADRs) and ensures the safety of medicines through post-marketing surveillance. Implements strategies to mitigate risks associated with drug use.
3. Clinical Trial Oversight: Grants permissions for conducting clinical trials in India. Ensures ethical and scientific standards are maintained during trials.
4. Quality Control: Inspects manufacturing facilities to ensure good manufacturing practices (GMP) are followed. Regulates the import and export of drugs and medical devices to ensure quality standards.
5. Licensing: Issues licenses for manufacturing, importing, and marketing drugs and medical devices.
6. Policy Development: Advises the government on drug regulation policies and amendments to existing laws. Collaborates with international regulatory agencies to align with global standards.
7. Combatting Drug Abuse: Regulates and monitors narcotics and psychotropic substances to prevent misuse.
8. Training and Capacity Building: Provides training to state drug regulatory authorities and other stakeholders to strengthen the regulatory framework.
Control Drugs Standard Control Organization (Cdsco): In India, the Central Drugs Standard Control Organization (CDSCO), which is supervised by the Ministry of Health and Family Welfare's Directorate General of Health Services, monitors and controls the production, distribution, and retailing of pharmaceuticals and cosmetics. It operates through a number of partnerships and bodies, including Central Drug Laboratories, the Drugs Consultative Committee, and the Drug Technical Advisory Board. In order to maintain uniformity in the administration of the Drugs and Cosmetics Act, CDSCO oversees the approval procedures for new medications, conducts clinical trials, creates prescription regulations, and keeps an eye on drug efficacy. India's primary regulatory body for medications, medical equipment, and cosmetics is the Central Drugs Standard Control Organization (CDSCO). Itfunctions under the Ministry of Health and Family Welfare's Directorate General of Health Services. throughout order to promote public health and safety, CDSCO is entrusted with guaranteeing the quality, safety, and efficacy of medical products throughout the nation.
Function:
1. Drug and Medical Device Regulation: Gives new medications and medical equipment permission to be marketed in India. guarantees adherence to the 1940 Drugs and Cosmetics Act and its implementing regulations.
2. Clinical Trials Oversight: Authorizes clinical trials to be carried out in India. oversees trials to make sure that moral and scientific guidelines are followed.
3. Pharmacovigilance: Keeps an eye on adverse drug reactions (ADRs) to guarantee the safety of pharmaceuticals.
4. Narcotics Regulation: Controls the
manufacture, distribution, and use of narcotics and psychotropic substances to prevent misuse.
5. Policy and Collaboration: Develops
guidelines and policies for drug and medical device regulation.Collaborates with international regulatory agencies to harmonize standards and practices.
ABBREVIATED:
The FDA receives the Abbreviated New Drug Application (ANDA) when it wants to approve generic medications. It permits a producer to create a generic version of a medication that has already received FDA approval after the exclusivity or patent period has passed. By proving that the generic medication is bioequivalent to the name-brand medication, an ANDA guarantees that the active ingredient, dosage form, mode of administration, strength, and performance characteristics are all the same. Comprehensive clinical trials are not necessary for this application; nonetheless, a number of investigations, including bioavailability, are required to demonstrate the drug's quality, safety, and efficacy. AANDA is submitted to regulatory bodies (such as the FDA in the US) in order to obtain permission to market a generic version of a reference medication that has already received approval. An Abbreviated New Drug Application (ANDA) is a submission made to the U.S. Food and Drug Administration (FDA) to seek approval for a generic drug. Unlike a New Drug Application (NDA), an ANDA does not require the applicant to submit extensive preclinical and clinical trial data. Instead, it demonstrates that the generic product is equivalent to an already-approved brand-name drug (reference listed drug or RLD).
Functions:
pharmaceutical companies to market generic drugs that are therapeutically equivalent to their brand-name counterparts. Ensures that the generic drug meets the same standards of quality, strength, dosage form, and route of administration as the RLD.
2. Bioequivalence Evaluation: Confirms that the generic drug delivers the same active ingredient to the body at the same rate and extent as the brand-name drug. Requires pharmacokinetic studies to demonstrate bioequivalence.
3. Cost Reduction: Facilitates the introduction of affordable alternatives to expensive brand-name drugs, increasing patient access to medications.
4. Regulatory Compliance: Ensures that the manufacturing, labelling, and packaging of the generic drug comply with FDA regulations and Good Manufacturing Practices (GMP).
5. Post-Approval Monitoring: Requires
manufacturers to monitor the safety and efficacy of the generic drug post-approval through adverse event reporting and periodic updates to the FDA.
6. Patent Certification and Exclusivity: Includes certification regarding patents associated with the RLD to avoid patent infringement. Grants market exclusivity for the first generic approved under certain circumstances (e.g., 180-day exclusivity for first-to-file applications).
New Drug Application (Nda): A crucial and complex step in introducing a new pharmaceutical product to the US market is the new drug application (NDA) procedure. It starts with a thorough preclinical testing process that includes both laboratory and animal tests to determine the safety profile and possible efficacy of the medicine. The sponsor submits an Investigation New Drugs (IND) application to the FDA after successful preclinical results, including comprehensive preclinical study data and a proposed clinical trial protocol. Phase I–III clinical studies use human participants to evaluate the drug's efficacy, safety, and ideal dosage, among other factors. To protect participant welfare and the accuracy of the data gathered, these trials are carried out in accordance with stringent ethical and legal requirements.
Function:
1) Predictive analytics using artificial intelligence (AI) and machine learning (ML): These technologies are being utilized to forecast patient outcomes and the course of diseases, which enables improved trial designs and more effective use of available resources.
2) Virtual and Decentralized Clinical Trials Remote Monitoring and Data Collection: Decentralized trials make trials more accessible and increase participant diversity by enabling patients to participate from home through the use of wearable technology, telemedicine, and smartphone apps.
3) Evidence (RWE) and Real-World Data (RWD) Integration of Real-World Data: Research is incorporating data from wearable technology, claims, and electronic health records (EHR). This supplements information from clinical trials by offering insights on how therapies function in actual environments.
4) Flexible and Adaptable Trial Plans Adaptive clinical trials: These enhance patient safety and resource efficiency by enabling researchers to make real-time adjustments to the study based on interim data, such as changing dosage or discontinuing unsuccessful therapies.
5) Transparency and Data Security Blockchain for Data Integrity: Blockchain technology protects patient privacy, increases transparency, and fosters trust while providing safe means of sharing and storing data.
FUTURE PROSPECTS [4]: Clinical research has an exciting future ahead of it thanks to new approaches and developing technologies that should increase speed, accuracy, and inclusion. The following are some key areas influencing clinical research going forward:
1) Predictive analytics using artificial intelligence (AI) and machine learning (ML): These technologies are being utilized to forecast patient outcomes and the course of diseases, which enables improved trial designs and more effective use of available resources.
2) Virtual and Decentralized Clinical Trials Remote Monitoring and Data Collection: Decentralized trials make trials more accessible and increase participant diversity by enabling patients to participate from home through the use of wearable technology, telemedicine, and smartphone apps.
3) Evidence (RWE) and Real-World Data (RWD) Integration of Real-World Data: Research is incorporating data from wearable technology, claims, and electronic health records (EHR). This supplements information from clinical trials by offering insights on how therapies function in actual environments.
4) Flexible and Adaptable Trial Plans Adaptive clinical trials: These enhance patient safety and resource efficiency by enabling researchers to make real-time adjustments to the study based on interim data, such as changing dosage or discontinuing unsuccessful therapies.
5) Transparency and Data Security Blockchain for Data Integrity: Blockchain technology protects patient privacy, increases transparency, and fosters trust while providing safe means of sharing and storing data.
Current Trends:
The field of clinical research is witnessing a number of significant trends driven by advancements in technology, regulatory adaptations, and a growing emphasis on patient centered approaches. Here are the main trends shaping clinical research today:
1. Decentralized Clinical Trials (DCTs): Remote Participation: Decentralized trials enable patients to participate from home through telemedicine, mobile applications, and wearable devices. This approach reduces the need for in-person visits and enhances accessibility, particularly for participants in remote or underserved areas.
2. Personalized and Precision Medicine:
Biomarker-Based Trials: Many studies are now tailored to specific patient subgroups based on genetic, molecular, or other biomarkers, leading to more targeted and potentially more effective treatments.
3. Enhanced Data Security and Privacy: Blockchain Technology: Some researchers are exploring blockchain as a secure, transparent way to manage patient data, improve data traceability, and maintain privacy. Compliance with Data Regulations: Increased focus on compliance with data protection regulations, such as GDPR and HIPAA, is leading to improved data management practices and patient trust.
4. Accelerated Drug Approval and Regulatory Adaptations: Fast-Track and Breakthrough Therapy Designations: Regulatory agencies are creating pathways that accelerate drug approval for therapies targeting unmet medical needs, allowing patients faster access to potentially life-saving treatments.
5. Increased Use of Data Sharing and Open Science: Collaborative Data Sharing: There’s an increasing push for data transparency and open science, with researchers sharing data across studies and institutions. This allows for greater reproducibility, validation, and new discoveries.
CONCLUSION: In summary, by tracking, evaluating, and averting negative effects, pharmacovigilance is essential to guaranteeing the safety of medications. To identify and manage possible dangers related to drugs and ultimately improve patient and public health, constant vigilance and strong reporting mechanisms are crucial. Continuous improvement in pharmacovigilance techniques will be essential to preserving the equilibrium between therapeutic advantages and safety considerations as the pharmaceutical industry changes. Healthcare providers and regulatory agencies can improve patient safety and aid in the ongoing development of medication therapy by methodically tracking and evaluating adverse drug reactions. But problems like underreporting and poor data quality still exist, highlighting the necessity of continuous efforts to improve pharmacovigilance systems around the world. In order to maintain a strong and proactive pharmacovigilance framework going ahead, cooperation between regulatory bodies, the pharmaceutical sector, and healthcare practitioners is still crucial.
ACKNOWLEDGEMENT:
The authors thankfully acknowledge the support provided by Kasturi Shikshan Sanstha’s College of Pharmacy, Maharashtra, India for carrying out the following review article.
REFERENCES


Sonawane Diksha*, Kandalkar Nilam, Gangawane Khushi, Bhandalkar Gauri, Sonawane Kajol, A Review Article of Clinical Research Studies and Pharmacovigilance for Healthcare, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 2, 1842-1851. https://doi.org/10.5281/zenodo.14910813
 10.5281/zenodo.14910813
10.5281/zenodo.14910813
