Pratibhatai Pawar College of Pharmacy Wadala Mhadev Shrirampur
The purpose of this evaluation was to enhance the solubility and bioavailability of poorly soluble medications through a variety of methods such as physical, chemical, and other modifications or techniques. Solubility refers to the maximum amount of solute that can dissolve in a specific amount of solvent or solution at a given temperature. Achieving the desired concentration of a drug in systemic circulation is crucial for the pharmacological response to be effective. Poor aqueous solubility can significantly impact drug efficacy and may lead to side effects. Various techniques are employed to improve aqueous solubility, which can ultimately enhance efficiency and reduce side effects for certain drugs. These techniques are applicable to parenteral, topical, and oral solutions. Physical modification techniques such as media milling/nanocrystal technology, cryogenic technology, supercritical fluid processes, crystal habit modification, complexation, micellar technologies, chemical modifications, and other methods like co-crystallization, co-solvency, and hydrotrophy are utilized to enhance the solubility of highly soluble drugs such as dolargin, loperamide, tubocurarine, doxorubicin, ibuprofen, griseofulvin, diazepam, naproxen, carbamazepine, nifedipine, and phytosterol.
The concept of solubility refers to the maximum amount of solute that can be dissolved in a given quantity of solvent or solution at a specific temperature. In simpler terms, solubility can also be described as the ability of one substance to form a solution with another substance. The substance that is being dissolved is known as the solute, while the liquid in which the solute dissolves is referred to as the solvent. Together, they create a solution. The act of dissolving the solute into the solvent is called solution or hydration, if the solvent happens to be water. More than 90% of drugs are administered orally, and the absorption and bioavailability of these drugs depend heavily on their solubility in water. Since 1995, over 90% of approved drugs have been found to have poor solubility. In fact, around 40% of new chemical entities identified in screening programs by pharmaceutical companies have low water solubility. This poses a significant challenge in delivering these drugs orally, as their absorption and bioavailability in humans need to be sufficient and consistent. The World Health Organization assigns BCS classifications to orally administered drugs based on publicly available data. Out of the 130 drugs on the WHO list, 61 could be classified with certainty. Among these, 84?long to class I (highly soluble, highly permeable), 17% to class II (poorly soluble, highly permeable), 39% to class III (highly soluble, poorly permeable), and 10% to class IV (poorly soluble, poorly permeable). The absorption rate and extent of class II and class IV compounds are greatly influenced by their bioavailability, which is ultimately determined by their solubility. Therefore, enhancing solubility is a crucial factor to consider in the formulation development of orally administered drugs with poor aqueous solubility. Solubility refers to the ability of a substance (solute) to dissolve in a solvent and is an important physical property to consider in drug development. (1) The phenomenon of solubility enhancement remains a topic of extensive discussion, yet it is not completely resolved. It poses a significant challenge for researchers in the field of formulation design and development. Solubility and dissolution are fundamental concepts in both physical and chemical sciences, with important biopharmaceutical and pharmacokinetic implications for medication treatment. Consequently, over 40% of new chemical compounds fail to progress in the drug development process due to suboptimal biopharmaceutical properties, such as absorption rate and distribution rate. As per IUPAC, solubility can be defined as the analytical composition of a saturated solution, expressed in terms of the proportion of a designated solute in a designated solvent. This solubility can be quantified using various measures such as concentration, molality, mole fraction, and mole ratio. The solubility of a poorly water-soluble drug presents a common challenge in screening studies of New Chemical Entities (NCE) and in the design and development of formulations. Various methodologies can be developed to enhance the bioavailability of such drugs. When administered orally, these drugs are completely absorbed but exhibit limited solubility in the gastric medium and good bioavailability. However, the bioavailability is influenced by factors such as drug permeability through lipophilic membranes. Analytically measuring solubility at low concentrations is therefore difficult. To facilitate rapid and efficient formulation development, a solubility classification was introduced to select an appropriate formulation system for highly active compounds with good permeability. In August 2000, the U.S. FDA issued a Guidance for Industry that covered the Biopharmaceutical Classification System (BCS). The BCS is a scientific framework for classifying drug substances based on their equilibrium aqueous solubility and intestinal permeability. When combined with the in vitro dissolution characteristics of a drug product, the BCS considers three major factors: solubility, dissolution rate, and intestinal permeability. These factors govern the rate and extent of oral drug absorption for immediate release of solid oral dosage forms. The BCS defines four classes of drug substances based on their solubility and permeability characteristics. (2
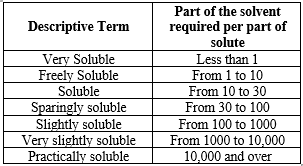
Table No.:1 USP and BP solubility Criteria
Solubility and its Types:
The ability of a solute, which can be a solid, liquid, or gas, to dissolve in a solvent, which can also be a solid, liquid, or gas, and form a uniform solution is known as solubility. The solubility of a substance is primarily influenced by the type of solvent employed, along with temperature and pressure. The saturation concentration, which is the point at which adding more solute does not raise its concentration in the solution, is used to quantify the extent of solubility in a specific solvent.
Absolute/Intrinsic Solubility:
The highest quantity of solute that can be dissolved in a specific solvent under standard conditions of temperature, pressure, and pH is referred to as absolute or intrinsic solubility. This characteristic is considered to be unchanging.
Saturated Solubility:
The maximum quantity of solute that can be dissolved in a specific solvent until it reaches its saturation point. Any extra solute will not be able to dissolve in the solvent. (3)
Importance of Solubility:
Oral ingestion is widely used and considered the most convenient method of drug delivery due to its ease of administration, high patient compliance, cost-effectiveness, minimal sterility requirements, and flexibility in dosage form design. Consequently, many generic drug companies prioritise the production of bioequivalent oral drug products. However, the main challenge in designing oral dosage forms lies in their limited bioavailability. Several factors, including aqueous solubility, drug permeability, dissolution rate, first-pass metabolism, presystolic metabolism, and susceptibility to efflux mechanisms, influence oral bioavailability. Poor solubility and low permeability are the most common causes of low oral bioavailability. Solubility is a critical factor in various dosage forms, including parenteral formulations. It is a key parameter in achieving the desired drug concentration in the systemic circulation to elicit the necessary pharmacological response. Drugs with poor water solubility often necessitate high doses to achieve therapeutic plasma concentrations following oral administration. The low aqueous solubility of drugs poses a significant challenge in the formulation development of both new chemical entities and generic drugs. For a drug to be absorbed, it must be in the form of an aqueous solution at the absorption site. Water is typically the preferred solvent for liquid pharmaceutical formulations. The majority of drugs are weakly acidic or weakly basic, further contributing to their poor aqueous solubility. Over 40% of new chemical entities (NCEs) developed in the pharmaceutical industry are practically insoluble in water. These drugs, which have low solubility, result in slow drug absorption, leading to inadequate and inconsistent bioavailability and gastrointestinal mucosal toxicity. For drugs administered orally, solubility is the most crucial factor in achieving the desired concentration in the systemic circulation for a pharmacological response. The problem of solubility poses a significant challenge for formulation scientists. Improving the solubility of drugs, and consequently their oral bioavailability, remains one of the most difficult aspects of the drug development process, particularly for oral drug delivery systems. There are various approaches available and documented in literature to enhance the solubility of poorly water-soluble drugs. The selection of techniques is based on factors such as the properties of the drug being considered, the nature of excipients to be used, and the intended dosage form. The inadequate solubility and slow dissolution rate of poorly water-soluble medications in the aqueous gastrointestinal fluids often result in insufficient bioavailability. Particularly for class II substances (low solubility and high permeability) as per the Biopharmaceutics Classification System (BCS), enhancing the solubility and dissolution rate of the drug in the gastrointestinal fluids can improve bioavailability. In the case of BCS class II drugs, the rate-limiting step is the release of the drug from the dosage form and its solubility in the gastric fluid, rather than absorption. Therefore, increasing solubility ultimately boosts bioavailability for BCS class II drugs.(4) The solubility of a drug plays a crucial role in various dosage forms, including parenteral formulations. Achieving the necessary drug concentration in the bloodstream and the desired pharmacological effect heavily relies on solubility (5,6). In some cases, when medications with poor water solubility are taken orally, high dosages may be necessary to achieve therapeutic plasma levels. The challenge lies in the low water solubility of these medications, which also affects the formulation of new chemical compounds and the development of genetic material (5,7). Water serves as the optimal solvent for liquid medicinal compositions due to its ability to facilitate the presence of substances that require absorption at the absorption site in the form of an aqueous solution. The majority of medications exhibit low aqueous solubility and tend to be either weakly basic or mildly acidic (8).
Solvent:
The primary element that comprises a solution and has the ability to dissolve another substance, resulting in a uniformly dispersed mixture at the molecular level.
Solute:
The solubility of a solute refers to the maximum amount of solute that can dissolve at a given temperature. (1)
Solubility:
Solubility can be described both qualitatively and quantitatively. In quantitative terms, solubility refers to the concentration of solute in a saturated solution at a specific temperature. On the other hand, qualitatively, solubility refers to the spontaneous interaction between two substances to create a uniform molecular dispersion. (9,10) The solubility refers to the quantity of substance that dissolves in a solution to achieve equilibrium under constant pressure and temperature, resulting in the formation of a saturated solution (11). The solubility of a substance is an important factor to consider. By examining the modified Noyes-Whitney equation, we can gain insights on enhancing the dissolution rate of compounds with low solubility. This, in turn, helps to reduce the constraints on the oral bioavailability of such substances (12).
dC/dt = AD (Cs - C)/h
Where,
dC/dt is the rate of dissolution, A is the surface area available for dissolution, D is the diffusion coefficient of the compound, Cs is the solubility of the compound in a dissolution medium, C is the concentration of drug present in the medium at a time t and h is the thickness of the diffusion boundary layer adjacent to the surface of the dissolving compound (13).
Biopharmaceutical Classification System (BCS)
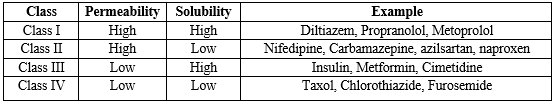
Amidon and colleagues created the Biopharmaceutical Classification System (BCS), which categorizes drugs based on three crucial factors affecting absorption: solubility, dissolution rate, and permeability. The BCS places drugs into one of four groups, as outlined in Table 2. (3)
Class I Drugs:
The drugs belonging to this class demonstrate a high absorption rate and a high dissolution rate. These compounds are efficiently absorbed by the body, and their absorption rate is typically higher than the excretion rate. Examples of Class I drugs include Diltiazem, Verapamil, and Propanol.
Class II Drugs:
In contrast, the drugs in this class have a high permeability but a low dissolution rate. The rate at which the drug dissolves become a limiting factor for absorption. Examples of Class II drugs include Nifedipine and Phenytoin.
Class III Drugs:
For Class III drugs, the rate of permeability becomes the limiting step for absorption. These drugs also exhibit variable absorption due to their high solubility but low permeability. Examples of Class III drugs include Insulin and Ranitidine.
Class IV Drugs:
Lastly, Class IV drugs have both low solubility and low permeability, making them poorly absorbed by the body. Both solubility and permeability act as rate-limiting steps for absorption. Examples of Class IV drugs include Thiazide and Taxol.
Note:
It is important to mention that Class II drugs, which have low solubility but high permeability, are preferred for solubility enhancement (14).

Fig.no.1: BCS Classification
Table No.2: BCS Classification of Drug
Process of Solubilization:
The solubilization process entails the disruption of inter-ionic or intermolecular bonds within the solute, resulting in the dispersion of solvent molecules to create room for the solute. This process involves the interaction between the solvent and the solute molecule or ion. During the occurrence of solubilization, the bonds of the solute break down, leading to the formation of visible gaps. The solvent contains an integrated freed solid molecule (15).

Fig.no.2: Process of Solubilization
Method To Enhance Solubility:
Chemical Modifications:
Physical Modifications:
a. Particle size reduction
i. Micronization
ii. Nanosuspension
b. Modification of the crystal habit
i. Amorphous form
ii. Polymorphs
c. Complexation
Chemical Modifications:
The formation of salts is a widely used and efficient technique to enhance the solubility and dissolution rates of acidic and basic pharmaceuticals. When an acidic or basic drug is converted into a salt form, it typically exhibits greater solubility compared to the original drug. Examples include Aspirin, Theophylline, and Barbiturates.
Let's discuss the concept of enhancing a drug's solubility by introducing a water-miscible solvent known as a co-solvent. In essence, combining a weak electrolyte with a co-solvent results in improved water solubility by decreasing interfacial tension. Examples of co-solvents include ethanol, sorbitol, PEG, glycerin, and others.
The solubility of a solute in water is enhanced by the presence of a significant quantity of additives, such as urea and sodium salicylate. This leads to an increase in the solubility of the solute.
The solubility of a poorly water-soluble drug can be enhanced through a process involving the addition of surface-active agents with higher HLB values, such as Polyoxyethylene fatty Acid (tween), ester polyoxyethylene mono alkyl sorbitan ether (BRIJ & MYRJ), and others.
Physical Modifications:
A. Particle size reduction
1. Micronization
The process involves the conversion of large particles into smaller particles of micron size. The reduction in particle size leads to an increase in surface area, which ultimately enhances the dissolution rate. In simpler terms, a smaller particle size indirectly improves bioavailability and solubility, as well as absorption due to greater exposure to the surface area. However, excessive reduction in particle size can result in the development of surface charges, leading to reduced flow ability and the formation of particle agglomerates. Examples of substances affected by this include Grisefulvin, aspirin, and Spironolactone.
B. Modification of crystal habbit
1. Amorphous form:
The solid in this state lacks a fixed internal structure, resembling the randomness of a liquid. This form, known as an amorphous solid, is created through rapid precipitation, lyophilization, and other methods. Amorphous solids are less stable than their isotropic counterparts, lacking a sharp melting point and exhibiting high solubility. The order of dissolution for various solid forms of a drug should be considered. Amorphous >Metastable polymorph >Stable polymorph
2. Polymorph
When a substance is found in multiple crystalline species with different internal lattice structures, these different forms are referred to as polymorphs, and the phenomenon is known as polymorphism. This occurrence is typically observed in organic compounds, as inorganic compounds are usually characterized by a single crystal system. For example, chloramphenicol Palmitate has three polymorphs: A, B, and C. Form B exhibits the highest bioavailability, while Form A is inactive. Physical properties such as solubility, melting point, density, crystal shape, and optical and electrical properties often vary between different polymorphs. The stable polymorph represents the lowest Gibbs energy, highest melting point, and greatest stability. The other polymorphs are considered metastable forms, which have higher Gibbs energy, lower melting points, and increased aqueous solubility. Due to their higher energy levels, metastable forms tend to convert to the stable form over time due to thermodynamic tendencies. It is important to note that a metastable form should not be mistaken for an unstable form, as it can remain stable for years if kept dry. Various methods can be used to analyze polymorphs, including crystallography, DSC (differential scanning calorimetry), optical microscopy, SEM (scanning electron microscopy), and TGA (thermogravimetric analysis) (14).
Factors Affecting Solubility:
Molecular Size:
The solubility of a drug is influenced by its molecular size. A larger molecule or higher molecular weight results in lower solubility. Larger molecules pose a challenge in being surrounded by solvent molecules for solvation. In organic compounds, an increase in carbon branching enhances solubility by reducing the size or volume of the molecule, thus facilitating solvation with solvent molecules (16).
Nature of the Solute and Solvent:
At room temperature, water can dissolve only 1 gram of lead (II) chloride per 100 grams of water, whereas 200 grams of zinc chloride can be dissolved. The significant contrast in solubilities between these two substances can be attributed to variations in their properties (17).
Temperature:
The solubility of a substance is influenced by temperature. When energy is absorbed during the solution process, an increase in temperature leads to an increase in solubility. Conversely, if energy is released during the solution process, solubility decreases as temperature rises (18). In most cases, raising the temperature of a solution enhances the solubility of a solid solute. However, some solid solutes exhibit lower solubility in warm solutions. Additionally, for all gases, solubility decreases as the solution temperature increases (17).
Pressure:
An elevation in pressure enhances the solubility of gaseous solutes, while a reduction in pressure diminishes their solubility. Conversely, alterations in pressure have minimal impact on the solubility of solid and liquid solutes (17).
Particle size:
The solubility is affected by the size of the solid particle due to the increase in surface area to volume ratio as the particle size decreases. This larger surface area enables a stronger interaction with the solvent. The impact of particle size on solubility can be explained (19).
Polarity:
The solubility of a substance is influenced by the polarity of both the solute and solvent molecules. In general, non-polar solute molecules will dissolve in non-polar solvents, while polar solute molecules will dissolve in polar solvents. Polar solute molecules possess both a positive and a negative end. When the solvent molecule is also polar, the positive ends of the solvent molecules will attract the negative ends of the solute molecules. This attractive force between the positive and negative ends is known as dipole-dipole interaction, which is a type of intermolecular force. Additionally, all molecules exhibit a weaker intermolecular force called London Dispersion forces. In this case, the positive nuclei of the solute molecule's atoms will attract the negative electrons of the solvent molecule's atoms. This allows the non-polar solvent to solvate the solute molecules (20).
Polymorph:
A solid possesses a rigid structure and a specific shape. While the appearance of a crystal formed from a particular substance may differ, the angles between its faces remain constant. Crystals are composed of atoms, ions, or molecules arranged in a regular geometric pattern that repeats in three dimensions, known as the unit cell. The ability of a substance to crystallize in multiple crystal forms is referred to as polymorphism. It is possible for all crystals to form different polymorphs. When the transition from one polymorph to another is reversible, it is termed enantiotropy. In a monotropic system, there exists a transition point above the melting points of both polymorphs, and the two forms cannot convert into each other without undergoing a phase transition. Polymorphs can have varying melting points, which in turn affects their solubilities. The solubilities of different polymorphs may differ by only 2-3 folds due to relatively small variations in free energy (21).
There are several methods accessible to enhance the solubility of drugs that have poor solubility. A few of the strategies to enhance solubility include:

Fig.no.3: Solubility Enhancement
The impact of pH changes in the gastrointestinal tract on the bioavailability of pharmaceuticals has been extensively documented. Drug absorption relies heavily on diffusion, which is influenced by the pH levels in different regions of the gastrointestinal tract, the drug's pKa, and permeability. These factors are not only affected by the surface area of the release region but also by the pH effects on drug ionization. Altering the pH can potentially dissolve poorly water-soluble drugs with protonatable (basic) or deprotonatable (acidic) parts of the molecule in water. While the significance of critical parameters such as salt selection and pH adjustment has been emphasized in pre-formulation, the use of pH-altering excipients in drug delivery systems is also highly beneficial. pH adjustment can be applied to both oral and parenteral administration methods.
Due to its strong buffering properties, the poorly soluble drug may precipitate upon intravenous administration when the pH ranges between 7.2 – 7.4. It is crucial to evaluate the buffer capacity and tolerance of the chosen pH to determine the feasibility of this method. The pH levels in the stomach range from 1 to 2, while in the duodenum it is between 5-7.5. Consequently, the solubility of the drug may be affected as it travels through the intestines during oral administration. By incorporating solubilized excipients that elevate the pH within a dosage form (such as tablets or capsules) above the pKa of weakly acidic drugs, the solubility of the drug can be enhanced. Similarly, excipients acting as alkalizing agents can boost the solubility of weakly basic drugs (23,24). After adjusting the pH, ionizable compounds (which can be acids, bases, or zwitterionic) become stable and soluble. This adjustment can also be effective for both crystalline and lipophilic compounds that have low solubility. The bioavailability of a compound can be enhanced if it precipitates upon dilution in a fine or amorphous form. This is because it creates a higher concentration gradient and a larger surface area for dissolution. In cases where the drug precipitates into poorly soluble forms, this method can be beneficial. If the particles that need to be dissolved are not able to quickly re-dissolve, the bioavailability may not be adequately enhanced. By increasing the solubility of the poorly soluble drug in comparison to water alone, there is a possibility of increasing the fraction of orally absorbed drug if the compounds can permeate through the epithelium. pH adjustment is often used in conjunction with co-solvents to further enhance the solubility of the poorly soluble drug.
This method is commonly employed in surveys because pre-clinical pH adjustment is an effective way to evaluate the effectiveness of poorly soluble drugs. It is favored for its widespread applicability and straightforward nature. The benefits of this approach include ease of formulation and analysis, quick production, minimal compound usage, and suitability for high-throughput assessments. However, there are notable drawbacks such as the risk of precipitation when diluted with aqueous solutions of differing pH levels, potentially leading to emboli when administered intravenously and causing variability and toxicity (both locally and systemically) when taken orally due to the use of non-physiological and extreme pH levels. However, if precipitation of the poorly soluble medication occurs uncontrollably after coming into contact with a pH level at which the medication is significantly less soluble (both orally and parenterally), the interpretation of the findings may be misleading. Another issue with this adjustment is that the chosen pH level may accelerate hydrolysis or catalyze other degradation mechanisms because a dissolved medication in an aqueous environment is often less chemically stable compared to a crystalline solid. Like all solubilized and dissolved systems, a dissolved medication in an aqueous environment is frequently less chemically stable compared to formulations in crystalline solid form. An example of a commercially available product that utilises pH adjustment is Phenytoin Injection (Epanutin® ready mixed, Pfizer) 50mg/ml, which contains co-solvents such as propylene glycol 40% and ethanol 10% (1.1 mmol Na+ per 5 ml ampoule) (22).
A microemulsion is a clear pre-concentrate that is isotropic, thermodynamically stable, and transparent or translucent. It consists of a mixture of oil, hydrophilic surfactant, and hydrophilic solvent that dissolves a poorly water-soluble drug. When in contact with water, the formulations disperse spontaneously to create a clear emulsion with very small and uniform oil droplets containing the solubilized poorly soluble drug. Microemulsions have been utilized to enhance the solubility of numerous drugs that are essentially insoluble in water, as well as for the inclusion of proteins for oral and parenteral administration (25,26). These uniform systems, which can be formulated with a wide range of surfactant concentrations and oil to water ratios, are all characterized by low viscosity. Surfactants, surfactant blends, and co-surfactants in micro-emulsions play a crucial role in enhancing the solubility of drugs formulated as micro-emulsions. An anhydrous system of micro-emulsions is known as a self-micro-emulsifying drug delivery system (SMEDDS) or micro-emulsion pre-concentrate. It consists of oil, surfactant, and co-surfactant, and can form an o/w micro-emulsion when dispersed in an aqueous phase under gentle agitation. The agitation necessary for self-emulsification is provided by stomach and intestinal motility (27). Surfactants such as polyoxymethylene surfactants like Brij 35 or sugar esters like sorbitan monooleate (Span 80), cationic or anionic surfactants like alkyl trimethylammonium bromide and sodium dodecyl sulfate, or zwitterionic surfactants such as phospholipids like lecithin (phosphatidylcholine) commercially available from soybean and eggs, can be non-ionic. Lecithins are particularly popular due to their excellent biocompatibility.
Due to the fluid nature of the product, most self-emulsifying systems are restricted to being administered in lipid-filled soft or hard-shelled gelatin capsules. It is important to consider the interaction between the capsule shell and the emulsion in order to prevent the hygroscopic contents from dehydrating or migrating into the capsule shell (28,29). Combinations of ionic and non-ionic surfactants have also been found to be effective. Micro-emulsion pre-concentrates remain clear even after dilution and typically contain a higher amount of water-soluble surfactant and a higher content of a hydrophilic solvent compared to macro-emulsion pre-concentrates. Due to the nature of the excipients, these formulations are only administered orally. Solubilization using micro-emulsion pre-concentrates is suitable for poorly soluble lipophilic compounds that have high solubility in oil and surfactant mixtures. The advantages of pre-concentrates include relatively easy manufacturing and not being dependent on digestion for drug release, resulting in optimal bioavailability and reproducibility without the need for co-administration of food (i.e. in the fasted state). However, a major disadvantage of micro-emulsions is their high concentration of surfactant/co-surfactant, which makes them unsuitable for intravenous administration. Diluting micro-emulsions below the critical micelle concentration of the surfactants could cause the drug to precipitate, but the fine particle size of the resulting precipitate would still enhance absorption. The tendency for the drug to precipitate upon dilution may be higher due to the dilution effect of the hydrophilic solvent. Formulations with high levels of synthetic surfactants may have poor tolerability in cases where long-term chronic administration is intended. Formulations containing It becomes increasingly difficult to validate multiple components. Micro-emulsion products, such as the poorly soluble compounds Aptivus® capsules (Boehringer Ingelheim GmBH) containing the HIV protease inhibitor tipranavir and Neoral® capsules (Novartis AG) containing the category defining immunosuppressant cyclosporine A, USP modified, serve as examples (22,30).
Self-emulsifying or self-micro emulsifying systems utilize the concept of in situ generation of emulsion within the gastrointestinal tract. The combination of oil, surfactant, co-surfactant, one or more hydrophilic solvents, and co-solvent creates a clear isotropic solution known as the self-emulsifying drug delivery system (SEDDS), without the need for an external phase (water). This solution forms fine o/w emulsions or micro-emulsions spontaneously upon dilution by the aqueous phase in the GIT, enhancing the dissolution and absorption of lipophilic drugs. The ease of emulsification is linked to the ability of water to penetrate the various liquid crystalline or gel phases that develop on the droplet's surface. One advantage of SEDDS in terms of scaling up and manufacturing is their spontaneous formation upon gentle mixing of components, remaining thermodynamically stable. However, drawbacks of this system include drug chemical instabilities and high surfactant concentrations. The high surfactant content in self-emulsifying formulations (30-60%) can cause irritation in the GIT. Most self-emulsifying systems are typically administered in lipid-filled soft or hard-shelled gelatin capsules due to the liquid nature of the product. It is important to consider the interaction between the capsule shell and the emulsion to prevent the hygroscopic contents from dehydrating or migrating into the capsule shell. Neoral® represents a selfmicroemulsifying drug delivery system (SMEDDS). Depending on the dosage, the relative bioavailability of cyclosporine A when administered as Neoral® can range from 174% to 239% compared to the bioavailability of cyclosporine A from Sandimmune®, the original formulation on the market. The size of emulsion droplets plays a significant role in influencing the bioavailability of drugs from emulsion formulations, as smaller droplet radii can increase plasma drug levels, partly due to direct lymphatic uptake. Given the high concentration of surfactants in SMEDDS, they are best suited for oral applications and may not be recommended for long-term use due to the potential risk of causing diarrhoea (22,31).
The pharmaceutical significance of a drug lies in its crystalline form, considering stability and bioavailability. Polymorphism, which refers to the presence of a drug substance in various. Variations in melting point, density, stability, and drug solubility can arise due to the escaping tendency of molecules from a specific crystalline structure. Typically, the most stable form of a drug is the one with the highest level of crystallinity, existing in various polymorphic forms with the lowest free energy content, resulting in the highest melting point and lowest solubility. By carefully managing the crystallization process, it is possible to deliberately produce amorphous or metastable forms of drugs with elevated free energy levels. While these forms offer increased solubility, they may face stability challenges unless stabilizers are included in the formulation to prevent crystal growth. A notable example highlighting the significance of polymorphism is the withdrawal of ritonavir (Norvir®) capsules from the market in 1998. This decision was made after a less soluble polymorph was discovered two years post-approval, leading to reduced drug bioavailability. This event underscored the pharmaceutical industry's awareness of the critical role of polymorphism and prompted the integration of polymorph screening as a standard practice in preformulation studies (22).
The bioavailability of poorly soluble drugs is frequently linked to the size of drug particles. Decreasing the particle size can enhance the dissolution characteristics of the drug, enabling a broader range of formulation methods and delivery technologies. The larger surface area resulting from reduced particle size allows for increased interaction with the solvent, leading to improved solubility. Traditional methods of particle size reduction, such as comminution and spray drying, rely on mechanical stress to break down the active compound. However, these mechanical forces, like milling and grinding, can subject the drug product to significant physical stress, potentially causing degradation. Thermal stress is also a concern during comminution and spray drying, especially when dealing with thermo-sensitive or unstable active compounds. Furthermore, these conventional methods often struggle to reduce the particle size of nearly insoluble drugs (<0>
In recent years, there has been a significant rise in the utilization of particle size reduction through supercritical fluid (SCF) processes, thanks to the emergence of novel nano-sizing and solubilization technology. The applications and technologies involving supercritical fluids have experienced explosive growth as well. For over a century, it has been recognized that supercritical fluids (SCFs), particularly carbon dioxide as the most commonly employed one, have the ability to dissolve nonvolatile solvents. These supercritical fluids are characterized by temperatures and pressures that surpass their critical temperature (Tc). SCFs, or supercritical fluids, have the unique ability to exhibit properties of both a liquid and a gas when they reach their critical pressure (Tp). This makes them a safe, environmentally friendly, and cost-effective option. The low operating conditions, such as temperature and pressure, make SCFs particularly attractive for pharmaceutical research. When SCFs are near their critical temperatures, they become highly compressible, meaning that even slight changes in pressure can significantly impact the density and mass transport characteristics of the fluid. These characteristics play a crucial role in determining the solvent power of the SCF. Above its critical temperature (Tc) and pressure (Pc), a SCF exists as a single phase. SCFs possess properties that are beneficial for product processing, as they exhibit intermediate characteristics between pure liquids and gases. This includes liquid-like density, gas-like compressibility and viscosity, as well as higher diffusivity compared to liquids. A supercritical fluid (SCF) is present as a single phase above its critical temperature (Tc) and pressure (Pc). SCFs possess properties that are advantageous for product processing as they exhibit characteristics that lie between those of pure liquids and gases (such as liquid-like density, gas-like compressibility and viscosity, and higher diffusivity compared to liquids). Close to critical temperatures, SCFs are highly compressible, enabling slight pressure adjustments to significantly impact the density and mass transport properties of a fluid, which largely influence its solvent capabilities. Additionally, the density, transport properties (like viscosity and diffusivity), and other physical attributes (such as dielectric constant and polarity) undergo significant changes with minor variations in operating temperature, pressure, or both near the critical points. Once drug particles are dissolved in SCF, they can be re-crystallized into much smaller particle sizes. The adaptability and accuracy provided by SCF processes facilitate the micronization of drug particles within specific size ranges, often reaching sub-micron levels. This capability allows for the precise adjustment of properties required for a particular application. The food industry has long utilized the unique processing capabilities of SCFs, and now these capabilities are being applied to pharmaceutical applications as well. Recent advancements in SCF processes have shown that they can be used to create nano-particulate suspensions with particle sizes ranging from 5 to 2,000nm. Pharmaceutical companies like Nektar Therapeutics and Lavipharm have specialized in particle engineering using SCF technologies to reduce particle size and enhance solubility. Supercritical solvents commonly used in these processes include carbon dioxide, nitrous oxide, ethylene, propylene, propane, n-pentane, ethanol, ammonia, and water. Various methods of SCF processing have been developed to address specific challenges, such as precipitation with compressed anti-solvents. The method of principal component analysis (PCA), the enhanced dispersion solution by supercritical fluids (SEDS), supercritical anti-solvents processes (SAS), and the aerosol supercritical extraction system (ASES) were utilized (22,34).
Inclusion complexes, also known as lipophilic drug-cyclodextrin complexes, can be easily formed by combining the drug and excipients together. This process leads to an improved solubility of the drug. Among various techniques to enhance solubility, inclusion complex formation has been specifically employed to enhance the aqueous solubility, dissolution rate, and bioavailability of poorly water soluble drugs. Cyclodextrins (CD) are cyclic oligosaccharides that are structurally related and have a polar cavity and hydrophilic external surface. Inclusion complexes are formed when a nonpolar molecule or region (known as the guest) is inserted into the cavity of another molecule or group of molecules (known as the host). Cyclodextrins are commonly used as host molecules. Cyclodextrins with 6, 7, and 8 D glucopyranosyl units connected to ? -1, 4 glycosidic linkages are referred to as ?, ?, ? cyclodextrins, respectively. In pharmaceutical formulations, derivatives of ?-cyclodextrin with increased water solubility, such as hydroxypropyl-?-cyclodextrin(HP-?-CD), are commonly used. Cyclodextrins have a donut-shaped ring structure consisting of glucose monomers. Hydrophilic cyclodextrins are safe in normal doses, while lipophilic ones may be toxic. Therefore, derivatives of natural cyclodextrins, such as methyl, hydroxypropyl, sulfoalkylated, and sulfated derivatives, which possess improved aqueous solubility, are preferred for pharmaceutical use. The ring structure of cyclodextrins has a hydrophilic exterior and a lipophilic core, allowing appropriately sized organic molecules to form noncovalent inclusion complexes. This results in increased aqueous solubility and chemical stability. The driving forces for complexation include the exclusion of high energy water from the cavity, the release of ring strain (particularly in the case of ? -CD), Vander Waals interactions, and hydrogen and hydrophobic bindings. Cyclodextrin complexes have demonstrated the ability to enhance the stability, wettability, and dissolution of the lipophilic insect repellent N, N-diethyl-m-toluamide (DEET), as well as the stability and photo stability of sunscreens. Due to their large molecular weights exceeding 1000Da, cyclodextrins are not expected to easily penetrate the skin. However, studies have reported varying effects on skin penetration when cyclodextrins are complexed with other substances. The solubilization achieved through complexation is a result of specific interactions, rather than changes in the overall properties of the solvent, as seen in other solubilizing systems such as co-solvents, emulsions, and pH adjustments. In a recent review conducted by Loftsson and Masson, it was suggested that the impact on skin penetration may be influenced by the concentration of cyclodextrins. The decrease in flux is typically observed at higher cyclodextrin concentrations, while lower cyclodextrin concentrations lead to an increase in flux. Since flux is directly related to the concentration of free drug, an increase in flux can be anticipated when the cyclodextrin concentration is adequate to complex only the drug that exceeds its solubility. The dissociation process is rapid, complete, and therefore predictable. Another notable advantage of the complexation technique is that certain commonly used complexing agents like hydroxypropyl beta cyclodextrin and sulfobutyl beta cyclodextrin are less toxic compared to other solubilizing agents such as surfactants and co-solvents. However, at higher cyclodextrin concentrations, the excess cyclodextrin is expected to complex with free drug, thereby reducing flux. Cyclodextrins have also been linked to enhancing skin penetration by extracting stratum corneum lipids. It is worth noting that most studies reporting cyclodextrin-mediated flux enhancement have utilized rodent model membranes, where lipid extraction is much simpler compared to human skin, suggesting that the penetration enhancement from cyclodextrin complexation may be overstated. Since the majority of complexes formed are 1:1 complexes of the AL type, diluting these complexes will not result in a solution that is supersaturated with respect to the substrate. This is particularly crucial for highly insoluble compounds that may precipitate upon injection when solubilized by other systems such as co-solvents. Shaker and colleagues recently determined that complexation with HP-?-CD did not impact the flux of cortisone through hairless mouse skin through either of the proposed mechanisms. The solubility enhancement application, CDs can also be used as membrane permeability enhancer and stabilizing agents. The permeability through biological membrane is enhanced by the presence of cyclodextrins. CDs can also be used as nasal permeation enhancers acting by interaction with nasal epithelium by modifying tight junction & lipid and protein content of the membrane, which enhances the permeation of the membrane. CDs can also be utilized as permeation enhancer in pulmonary drug delivery systems. Rifampicin is a so- called concentration-dependent antibiotic, the rate and extent of bacterial kill is related to the attainment of high maximum concentration relative to the minimal inhibitory concentration. The rifampicin-CD inclusion compound can improve the lung transport of drug whennebulized with compatible pulmonary deposition and achieve required concentration of drug in broncho-alveolar epitheium lining-fluid when administered as aerosolized solution. The enzymatic degradation of starch by cyclodextrin-glycosyl transferase (CGT) produces cyclic oligomers, Cyclodextrins. Cyclodextrins are non-reducing, crystalline, water soluble, cyclic, oligosaccharides. Solubility and oral bioavailability of Glipizide53, Rofecoxib54, Piroxicam55 and Carvedilol56 can be improved by using cyclodextrins inclusion complex. Solubilization by complexation is achieved through specific Interaction rather than changes in the bulk solvent properties, as seen in other solubilizing systems like co-solvents, emulsion, and pH adjustments, is the key factor in the rapid and quantitative dissociation observed with cyclodextrins (CDs). Masson highlighted the permeation enhancement property of poorly water-soluble drugs in the presence of CDs. These CDs act as permeation enhancers by facilitating the transport of the drug through the aqueous barrier that exists before the lipophilic surface of biological membranes. This dual characteristic of CDs, being both lipophilic and hydrophilic, contributes to their effectiveness. However, despite the advantages of complexation, there are some drawbacks. Firstly, the compound must be capable of forming complexes with the selected ligand. For compounds with very limited solubility, the enhancement of solubility may be minimal. Secondly, in the case of Ap-type complexes, dilution of the system can still lead to precipitation. This is also true for solubilization techniques involving combined methods such as complexation with pH adjustment. Lastly, the potential toxicity, regulatory concerns, and quality control issues associated with the presence of the ligand can complicate and increase the cost of the development process. (22,35)
The addition of a water miscible solvent, known as cosolvents, can frequently increase the solubility of a poorly water soluble drug . Co-solvents are mixtures of water and one or more water miscible solvents that are used to create a solution with enhanced solubility for compounds that are poorly soluble. This technique has been widely used historically because it is simple to produce and evaluate. Co-solvency has been utilized in various formulations, including solids and liquids. Examples of solvents used in co-solvent mixtures include PEG 300, propylene glycol, and ethanol. To increase the solubility and dissolution of meloxicam, solid binary systems with polyethylene glycol 6000 at various concentrations (5-40%) were employed. Co-solvent formulations of poorly soluble drugs can be administered orally or parenterally. For parenteral formulations, the addition of water or a dilution step with an aqueous media may be required to lower the solvent concentration prior to administration. Co-solvency techniques have also been used in spray freezing of liquid formulations, such as danazol with polyvinyl alcohol, poloxamer 407, and poly vinyl pyrrolidone K-15 in a micronized powder formulation. The pharmaceutical form is always liquid. A co-solvent approach may be suitable for poorly soluble compounds that are lipophilic or highly crystalline and have a high solubility in the solvent mixture. Co-solvents can significantly increase the solubility of poorly soluble compounds compared to their aqueous solubility alone. High drug concentrations of poorly soluble compounds can be effectively dissolved compared to other solubilization methods. Co-solvency, commonly used in various formulations, is particularly beneficial in parenteral dosage forms due to the irritating effects of surfactants and the low toxicity of many co-solvents. Propylene glycol, ethanol, glycerin, and polyethylene glycol are among the frequently used low toxicity co-solvents for parenteral applications. The use of co-solvents is a valuable technique for improving the solubility of poorly soluble drugs. Co-solvents are widely utilized due to their high solubilization capacity for poorly soluble drugs and their relatively low toxicity. They offer several advantages, such as simplicity and rapidity in formulation and production. However, like all excipients, the toxicity and tolerability associated with the level of solvent administered must be taken into consideration. Additionally, uncontrolled precipitation can occur when diluted with aqueous media, resulting in amorphous or crystalline precipitates of varying sizes. Many insoluble compounds are not suitable for co-solvents alone, especially for intravenous administration, as they are highly insoluble in water and do not readily re-dissolve after precipitation from the co-solvent mixture. This poses a potential risk for embolism and local adverse effects at the injection site. Furthermore, the chemical stability of insoluble drugs in solubilized forms is inferior to that in a crystalline state. Examples of co-solvent formulations include Nimodipine Intravenous Injection (Nimotop®, Bayer) and Digoxin Elixir Pediatric (Lanoxin®, GSK) (22).
The application of surfactants to enhance the dissolution performance of poorly soluble drug products has proven to be effective. By reducing surface tension, surfactants can enhance the dissolution of lipophilic drugs in aqueous solutions. Additionally, they can aid in stabilizing drug suspensions. Once the concentration of surfactants surpasses their critical micelle concentration (CMC), typically ranging from 0.05-0.10% for most surfactants, micelle formation occurs, trapping the drugs within the micelles. This phenomenon, known as micellization, generally leads to increased solubility of poorly soluble drugs. Commonly utilized non-ionic surfactants include polysorbates, polyoxyethylated castor oil, polyoxyethylated glycerides, lauroyl macro glycerides, and mono- and di-fatty acid esters of low molecular weight polyethylene glycols. Surfactants are also frequently employed to stabilize micro-emulsions and suspensions in which drugs are dissolved. Examples of poorly soluble compounds that benefit from Micellar solubilization include antidiabetic drugs such as gliclazide, glyburide, glimepiride, glipizide, repaglinide, pioglitazone, and rosiglitazone. Micelles are typically categorized into two groups.
a. Mixed Mecelles:
The micellar phase is the preferred phase for short chain analogues due to their chemical structure, temperature, and water content, whereas long chain phospholipids form bilayers when dispersed in water. Mixed micelles provide a hydrophobic core that allows for the dissolution of low soluble compounds. Both water and organic solvents can spontaneously form thermodynamically stable micellar solutions. The process of micelle formation is involved in this phenomenon. The critical micellar concentration (CMC) is the minimum solute concentration required for the occurrence of micelles. Additionally, micelles can only form at solution temperatures above the critical micellar temperature (CMT). These small colloidal aggregates, known as micelles, exist in rapid thermodynamic equilibrium with a measurable concentration of monomers. The size of micelles typically ranges from 5 to 10 nm, and their shape is ultimately determined by the chemical structure of the detergent. Micelles have the ability to solubilize host molecules, such as drugs, within their volume. However, the penetration of these molecules into the micelle is influenced by factors such as the inner space of the micelle, the hydrophobicity of the drug, and the charge of the incorporated molecule. The interaction between micelles and lipophilic drugs can result in the formation of mixed micelles (MM), also known as swollen micelles. The addition of substances like salt or alcohol can alter the degree of penetration into the micelle, a phenomenon referred to as co-solubilization. It is important to consider the potential toxic side effects of certain drugs on human cells, as well as their taste. The kinetics of micelles are driven by both rapid exchanges between micelles and monomers, as well as the dissolution and formation of new micelles. Despite the extreme disorder within the micelle interior and the extensive contact between water and methylene and methyl groups, swollen micelles are fluid systems that remain stable enough to be utilized as delivery systems for stable drugs.
b. Polmeric Micelles:
Amphiphilic polymers form nano-sized supra molecular core-shell structures, referred to as polymeric micelles, which are currently the subject of extensive research for drug delivery purposes. These micelles are considered to be potentially safer for injection compared to traditional solubilizing agents, allowing for higher doses of potent yet poorly water-soluble compounds. Polymeric micelles effectively solubilize essential but poorly water-soluble substances like amphotericin B, propofol, paclitaxel, and photosensitizers. The key to successful drug solubilization lies in the compatibility between the drug and the core of the polymeric micelle. Various materials such as Pluronics, poly (ethylene glycol) (PEG)-phospholipid conjugates, PEG-b-poly (ester)s, and PEG-b-poly (L-amino acid) are being explored for drug delivery applications. Polymeric micelles have the ability to circulate in the bloodstream for extended periods, evade the body's defense mechanisms, gradually release the drug, and exhibit a tendency to accumulate at disease sites like solid tumors. Additionally, polymeric micelles have shown promise in inhibiting p-glycoprotein in drug-resistant tumors, the gastrointestinal tract, and the blood-brain barrier, potentially offering a solution to drug resistance in cancer treatment, enhancing drug absorption in the gut and brain, and reducing the self-aggregation of polyene antibiotics - crucial drugs in combating severe systemic fungal infections. Ultimately, polymeric micelles have the potential to decrease the toxic effects of these drugs without compromising their antifungal properties (22,36).
Hydrotrophy refers to a process in which the addition of a significant amount of a second solute leads to an increase in the solubility of another solute in water. This increase in solubility is attributed to the presence of a large quantity of additives. The mechanism behind this improvement in solubility is closely related to complexation, which involves a weak interaction between hydrotropic agents such as sodium benzoate, sodium acetate, sodium alginate, urea, and poorly soluble drugs (2). The solute in question consists of alkali metal salts of various organic acids, while hydrotropic agents are ionic organic salts. Salts or additives that enhance solubility in a given solvent are referred to as "salting in" the solute, whereas salts that decrease solubility are known as "salting out" the solute. Certain salts with large anions or cations that are highly soluble in water lead to the phenomenon of "salting in" non-electrolytes, which is called "hydrotropism". Hydrotropic solutions do not exhibit colloidal properties and involve a weak interaction between the hydrotropic agent and the solute. The advantages of the Hydrotropic Solubilization Technique include its independence from pH, high selectivity, and the absence of the need for emulsification. It simply requires mixing the drug with the hydrotrope in water, without the need for chemical modification of hydrophobic drugs, the use of organic solvents, or the preparation of an emulsion system. The categorization of hydrotropes based on molecular structure poses a challenge due to the wide array of compounds that have been identified to display hydrotropic behavior. For instance, ethanol, aromatic alcohols like resorcinol, pyrogallol, catechol, aand b-naphthols, and salicylates, alkaloids such as caffeine and nicotine, ionic surfactants like diacids, SDS (sodium dodecyl sulfate), and dodecylated oxidibenzene are some specific examples. Aromatic hydrotropes with anionic head groups are the most extensively researched compounds due to their large number resulting from isomerism. The effective hydrotrope action of these compounds may be attributed to the availability of interactive pi orbitals. Hydrotropes with cationic hydrophilic groups, such as salts of aromatic amines like procaine hydrochloride, are rare. Apart from improving the solubilization of compounds in water, they are also known to impact surfactant aggregation leading to micelle formation, phase behavior of multi-component systems in terms of nano-dispersions and conductance percolation, clouding of surfactants and polymers, among other effects. Additional techniques that enhance the solubility of poorly water-soluble drugs include salt formation, alteration in the dielectric constant of the solvent, chemical modification of the drug, utilization of hydrates or solvates, application of soluble prodrugs, implementation of ultrasonic waves, and spherical crystallization (22).
Solid dispersion techniques involve dispersing a poorly soluble drug in a highly soluble solid hydrophilic matrix to improve drug dissolution. This method can result in eutectic or solid solution products. For example, a solid dispersion of carbamazepine in polyethylene glycol 4000 increased the dissolution rate and extent of the drug. The process includes loading a precipitation vessel with a solution of the drug and PEG4000 in acetone, then expanding it with supercritical CO2 to obtain solvent-free particles (22). Eutectic dispersions are homogeneous mixtures of crystalline or amorphous drugs in carriers, while solid solutions involve partial or complete solubility of the drug in the matrix. Despite the benefits of enhanced dissolution and simplicity, challenges in manufacturing, stability, and scale-up have hindered the widespread adoption of solid dispersion techniques. Originally proposed by Sekiguchi and Obi in the 1960s, solid dispersions are valuable for improving drug dissolution, absorption, and efficacy in dosage forms. Common hydrophilic carriers used in solid dispersions include polyvinylpyrrolidone, polyethylene glycols, Plasdone-S630, Tween-80, Docusate sodium, Myrj-52, Pluronic-F68, and Sodium Lauryl Sulphate. Solid dispersion can enhance the solubility of celecoxib, halofantrine1, and ritonavir by utilizing appropriate hydrophilic carriers (37).
A pharmaceutical Nano-suspension is a two-phase system comprising of drug particles in nano size that are stabilized by surfactants. It can be used for oral and topical administration as well as parenteral and pulmonary administration. Nano-suspension technology has emerged as a potential solution for effectively delivering hydrophobic drugs . This technology is particularly useful for drugs that have low solubility in both water and oils. The particle size distribution of solid particles in nano-suspensions is typically less than one. The average particle size of the micron ranges from 200 to 600 nm. Several techniques can be employed to prepare Nano-suspensions, such as Media Milling (Nanocrystals), High Pressure Homogenization in water (Dissocubes), High Pressure Homogenization in nonaqueous media (Nanopure), and a combination of Precipitation and High-Pressure Homogenization (Nanoedege) (38).
Patel Rajanikant and colleagues employed an innovative method to improve the dissolution of ibuprofen, a weakly acidic, non-steroidal anti-inflammatory drug, by developing a floating formulation. Ibuprofen exhibits high permeability in the stomach as it remains 99.9% un-ionized (pKa of Ibuprofen - 4.43, pH of gastric fluid - 1.2). Although it is mostly permeable through the stomach, its limited solubility prevents it from entering. The duration of systemic circulation and gastric emptying time ranges from 30 minutes to 2 hours. After this period, ibuprofen enters the small intestine where it becomes solubilized but cannot permeate through its membrane. To address this issue, it was decided to develop formulations that would remain in the stomach for more than 2 hours. This was necessary because the drug was not completely soluble within the initial 2-hour timeframe. To achieve complete dissolution in the stomach, a floating dosage form was designed. The floating ibuprofen granules were prepared using the fusion method. The formulation consisted of ibuprofen (200 mg divided into 50 mg and 150 mg), gelucire 44/14 (350 mg melted), and an additional 50 mg of ibuprofen. The ingredients were dispersed using a glass rod to ensure uniform distribution of the drug in the melted carrier. The remaining 150 mg of ibuprofen was then added to the melted gelucire 44/14, and this entire dispersion was added to the melted gelucire 43/01. In the optimized formulation, the granules remained floated for 3 hours and achieved 100% drug release within 150 minutes in the stomach region. During this time, the drug remained in a 99.9% unionized form and was absorbed into the systemic circulation (39).
Cryogenic methods have been developed to produce nano-structured amorphous drug particles with a high level of porosity under extremely low temperature conditions in order to improve the dissolution rate of drugs. Cryogenic innovations can be characterized by the type of injection device used (such as capillary, rotary, pneumatic, and ultrasonic nozzles), the positioning of the nozzle (above or below the liquid level), and the composition of the cryogenic liquid (including hydrofluoroalkanes, N2, Ar, O2, and organic solvents). Following cryogenic treatment, dry powder can be obtained through various drying techniques (such as spray freeze drying, atmospheric freeze drying, vacuum freeze drying, and lyophilization).
Nano crystallization refers to the process of reducing drug particles to a size range of 1-1000 nanometers. There are two primary approaches to producing nanocrystals: the "bottom-up" and "top-down" methods. The top-down methods, such as milling and high-pressure homogenization, involve starting with a macroscopic level material, typically a micron-sized powder, and gradually reducing its size. On the other hand, the bottom-up methods, such as precipitation and cryo-vacuum method, involve chemically composing nanoscale materials from atomic and molecular components.
Nanoscale particles can be generated through the wet milling technique. In ball mills, the reduction in particle size is accomplished by employing impact and attrition forces. The two commonly used models are the tumbling ball mill and the stirred media mill. However, this method poses challenges such as the deterioration of mill surfaces and the resulting contamination of the suspension.
In the process of high-pressure homogenization, the crystalline drug particles are dispersed in an aqueous solution and then passed through a narrow homogenization gap at a high velocity under high pressure. This homogenization can be carried out in water or in non-aqueous or water-reduced media. The particles are broken down through cavitations and shear forces. The static pressure on the liquid causes it to boil, forming gas bubbles. As the liquid exits the gap, the gas bubbles collapse under normal air pressure, creating shock waves that cause the crystals to collide and disintegrate. When working with temperature-sensitive materials, it is advisable to use a heat exchanger, as high-pressure homogenization can lead to an increase in sample temperature. The particle size achieved during the homogenization process is primarily influenced by the properties of the drug, the applied pressure, and the number of homogenization cycles.
In the precipitation technique, a weak solution is initially formed by dissolving the substance in a solvent. Subsequently, this solution containing the drug is introduced into water, which serves as an ineffective solvent. During the injection process, it is crucial to stir the water effectively to facilitate the precipitation of the substance in the form of nanocrystals. These nanocrystals can be separated from the solution through filtration and subsequently dried in the air.
D. Cryo vaccum method:
In the cryo-vacuum technique, the active component is initially dissolved in water to achieve a quasi-saturated solution. This technique relies on rapidly cooling a solvent by placing the solution in liquid nitrogen (-196 ºC), leading to a rapid increase in saturation levels due to reduced solubility and the formation of ice crystals once the temperature falls below 0 ºC. This results in quick nucleation of the dissolved substance around the ice crystals' edges. The solvent must be completely frozen before removing the vessel from the liquid nitrogen. Subsequently, the solvent is eliminated through sublimation in a lyophilization chamber where the temperature is maintained at a constant -22 ºC and the pressure is reduced to 10-2 mbar. Cryo-assisted sublimation enables the removal of the solvent without altering the size and structure of the produced particles, ensuring they remain crystalline. This method produces highly pure nanocrystals without the need for surfactants or harmful chemicals.
The Nanomorph technology involves the conversion of drug substances with low water-solubility from a coarse crystalline state into amorphous nanoparticles. A suspension of the drug substance in a solvent is introduced into a chamber, where it is quickly mixed with another solvent. This results in the immediate transformation of the drug substance suspension into a true molecular solution. The addition of an aqueous solution of a polymer causes the drug substance to precipitate. The polymer helps maintain the drug substance particles in their nanoparticulate state, preventing them from aggregating or growing. By utilizing conventional methods such as spray-drying, water redispersable dry powders can be produced from the nanosized dispersion. Through this technology, coarse crystalline drug substances can be converted into a nanodispersed amorphous state without the need for physical milling or grinding procedures, ultimately leading to the creation of amorphous nanoparticles (22).

Fig.no.4: Conventional approaches for solubility enhancement
FUTURE PROSPECTS:
Solid dispersions for poorly soluble drugs are expected to showcase significant advancements in technology related to preparation methods, scale-up processes, and the development of new techniques for better understanding the solid state structure. Future studies on solid dispersions should incorporate innovative methods to analyze drug solubility, molecular state, and its interaction with polymers to overcome challenges in designing new carriers that can prevent drug crystallization. It is crucial to extensively investigate the impact of storage conditions on drug properties, the carrier utilized, drug release profiles, and drug bioavailability. Additionally, substituting a hydrophobic carrier with a hydrophilic one may lead to a more controlled solid dispersion process. This approach could be easily adapted to achieve sustained drug release or modify solid state characteristics. The solid dispersion technique holds immense potential for future research and growth, potentially leading to the development of novel applications for oral drug delivery. The question arises whether solid dispersion technology can be universally applied to enhance the solubility of poorly water-soluble compounds. Currently, no single strategy for improving drug solubility in aqueous media is suitable for all drugs and dosing needs. However, assessing the stability of the amorphous state of solid dispersions remains a key area of interest that requires further exploration to fully leverage the benefits of this effective technique. Among the various techniques employed in industries to enhance the dissolution of poorly water-soluble drugs, solid dispersion appears to be the most versatile and applicable to a wide range of compounds and dosing requirements. A solution form of a drug allows for immediate absorption and is more efficiently absorbed compared to the same amount of drug in a solid dosage form like a tablet or capsule. The solubility of a drug is a crucial factor in determining its oral bioavailability, especially for poorly soluble drugs. The dissolution of the drug is the step that determines the rate of oral absorption for these drugs, which can subsequently impact their in vivo absorption. Currently, only 8-10% of new drug candidates possess both high solubility and permeability. Due to the solubility issues faced by many drugs, their bioavailability is affected, making solubility enhancement necessary. Various techniques mentioned above can now be employed to increase the solubility of poorly soluble drugs.
CONCLUSION
The drug's solubility plays a crucial role in determining both the drug's formulation and its therapeutic effectiveness, making it the most vital factor in formulation development. The dissolution of a drug is the determining factor in the oral absorption of poorly water-soluble drugs. Additionally, solubility is a fundamental requirement for formulating and developing different dosage forms of various drugs. The techniques mentioned above, either individually or in combination, can be utilized to improve the solubility of the drug. While all the techniques mentioned can enhance solubility, the selection of a method should be based on its effectiveness and the safety of the excipients used. For drugs administered orally, solubility is a crucial parameter that limits the rate at which they can achieve the desired concentration in the systemic circulation for a pharmacological response. Solubility can be increased through various techniques, resulting in a significant improvement in solubility. Due to the solubility issues faced by many drugs, their bioavailability is affected, making solubility enhancement essential. It is now possible to enhance the solubility of poorly soluble drugs using the techniques mentioned above. The dissolution of the drug is the key step in determining the rate of oral absorption, especially for poorly water-soluble drugs. Solubility is also a fundamental requirement for formulating and developing various dosage forms for different drugs. Solubility enhancement can be achieved through various techniques, resulting in a significant increase in solubility. The bioavailability of many drugs is negatively impacted by solubility issues, making solubility enhancement essential. It is now feasible to enhance the solubility of poorly soluble drugs using the aforementioned techniques. Solubility plays a crucial role in the oral bioavailability of poorly soluble drugs. The dissolution of a drug is the key step in determining the rate of oral absorption for poorly water-soluble drugs, ultimately affecting the drug's in vivo absorption. Currently, only 8% of new drug candidates exhibit both high solubility and permeability. Despite common methods like salt formation and particle size reduction being used to enhance drug dissolution rate, practical limitations exist, and the desired bioavailability enhancement may not always be achieved. Therefore, formulation approaches are being explored to improve the bioavailability of poorly water-soluble drugs.
REFERENCES


Kajal Santosh Kale, Shinde S. B., Solubility Enhancement Of Poorly Soluble Drug, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 7, 463-487. https://doi.org/10.5281/zenodo.12679933
 10.5281/zenodo.12679933
10.5281/zenodo.12679933
